




Prescribir un tratamiento es mucho más que extender una receta. En esto, nuestra lengua, a tenor de los que nos señala el Diccionario de la Real Academia Española (DRAE), no es especialmente clarificadora. Dice este diccionario que recetar es prescribir un medicamento, con expresión de sus dosis, preparación y uso. Es decir, trasladar una prescripción a quién debe dar cuenta y uso de ella. Prescribir es, según esta misma fuente, recetar, ordenar un remedio. Y el tan extendido uso del término indicación no nos saca del bucle ya que indicar se define como recetar remedios. Por tanto, recetar es ordenar a un determinado paciente que aplique aquello que sirve para producir un cambio favorable en las enfermedades, que es la definición del DRAE para la palabra remedio. Y si queremos ir a la esencia, es decir, a la palabra tratamiento, nos encontramos con que esta se define de manera general como conjunto de medios que se emplean para curar o aliviar una enfermedad. En resumen, cuando un médico prescribe lo que hace es indicar un medicamento del que se espera induzca un cambio favorable en el curso de la enfermedad del paciente. Pero esto es solo el final de un complejo proceso de deliberación con el paciente, en el que tras identificar de la mejor manera posible el curso previo de la enfermedad se toman decisiones sobre como intentar mejorar ese curso. La deliberación clínica implica intercambio de información, en cualquier caso, imprescindible para que la indicación terapéutica deje de ser una orden, para pasar a convertirse en un acuerdo que solemos llamar consentimiento informado.
La medicina es aplicar ciencia en ambiente de incertidumbre, mientras que la clínica es el arte que nos permite gestionar esa incertidumbre desde el fundamento científico. Esta gestión es siempre deliberativa y prudencial. Se orienta a la toma de la mejor decisión posible en el estricto interés del paciente. Los componentes de la decisión parten, en primer lugar, del conocimiento científico en todo lo que se refiere a los fundamentos de los hechos del enfermar. Sin conocer en profundidad el estado del arte, entendido como la permanente actualización de ese conocimiento, el médico no puede indicar, ya que la indicación sería, cuando menos, obsoleta, sino directamente maleficente. Y, desde luego, con esta carencia tampoco puede informar a su paciente, en ese escenario de deliberación clínica, de cara a un correcto consentimiento informado. Pero lo peor es que su posición médica pierde su fundamento, deja de ser consistente y se hace incompatible con la excelencia que debe siempre guiar el ejercicio médico.
En este número de la revista SEMERGEN se publica un importante original que ofrece datos preocupantes, en el ámbito del uso de los medicamentos biosimilares, respecto del conocimiento de causa que los médicos de atención primaria poseen a la hora de prescribir estos medicamentos1. En este original se establecen con precisión los conceptos más relevantes que todos debemos conocer, y que de una manera resumida pueden enumerarse de la siguiente forma: un biosimilar es una versión de un medicamento biológico; estas versiones, realizadas por fabricantes diferentes al que licenció la versión inicial u original, son factibles solo una vez caducada la correspondiente patente; de los biosimilares se espera una función superponible aunque no sean medicamentos idénticos; su mayor ventaja reside en el abaratamiento del producto biosimilar. Es clave entender que, al tratarse de medicamentos biológicos, los biosimilares nunca pueden ser considerados como genéricos. En los genéricos nos encontramos ante un medicamento idéntico al original, ya que no se trata de medicamentos biológicos y, por tanto, sí que cabe en este caso hacer copias estructurales idénticas. Este hecho implica una diferencia fundamental a la hora de gestionar el uso de estos medicamentos. Cuando un médico indica un principio activo del que existen varias marcas, originales y genéricas, la indicación está basada en ese principio activo, idéntico en las diferentes marcas. Por ello, un cambio de marca en la dispensación no implica cambio de indicación, refiriéndonos al resultado final del proceso de toma de decisiones que definíamos anteriormente. Esto es muy diferente en el caso de los biosimilares.
Los medicamentos biológicos tienen un proceso de investigación y de producción diferente al de las formas galénicas tradicionales. Los medicamentos biológicos son producidos a partir de seres vivos y se basan en estructuras complejas que buscan efectos ligados a su función biológica. Por las propias características del proceso de producción nunca un medicamento biológico es idéntico a otro, aunque pueda conseguirse una gran similitud. Los efectos de los medicamentos biológicos son variables y entre ellos debe tenerse siempre en cuenta la inmunogenicidad. Por todo ello, como se describe también en el original de Micó-Pérez et al.1, las agencias reguladoras, especialmente la Agencia Europea de Medicamentos (EMA), vienen realizando extraordinarios esfuerzos para garantizar la seguridad y eficacia de los biosimilares que llegan al mercado, estableciendo las debidas recomendaciones para su aprobación2, ocupándose también en publicar documentos informativos para profesionales3. Sin embargo, a pesar de la experiencia acumulada, todavía existe una alta variabilidad en los desarrollos clínicos de estos productos, por lo que el proceso de perfeccionamiento de estos procesos se mantiene en marcha4.
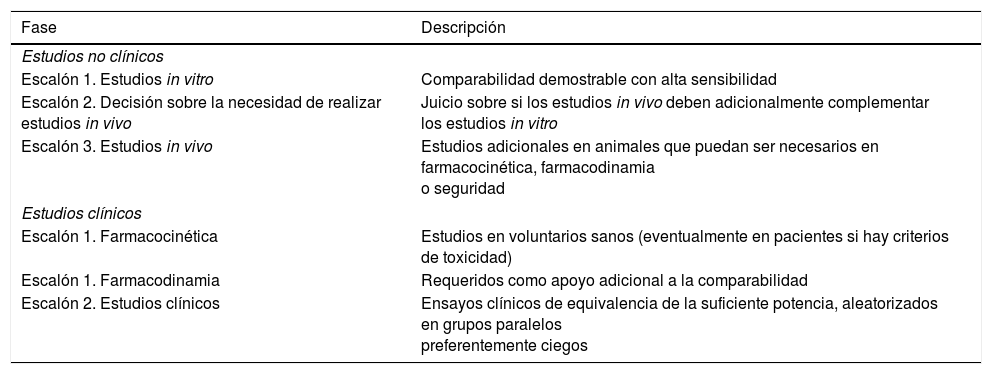
Los clínicos se han visto, en cierta medida, sorprendidos por la llegada de los biosimilares. Una potente razón para ello es la forma en la que estos medicamentos acceden al mercado. Desde hace décadas la innovación farmacológica ha llegado a través de un proceso consagrado para nuevos medicamentos, basada en la realización de ensayos clínicos (EC), cuya metodología está perfectamente establecida5 y regulada6. La publicación en revistas biomédicas de las sucesivas fases I a III de los ensayos, previas siempre a su aprobación, permiten conocer a los clínicos el fundamento científico de los nuevos medicamentos antes de su llegada al mercado. La aprobación final del nuevo producto resulta de un juicio institucional vinculante que está basado y es consecuente con la evidencia conocida y criticable derivada de los correspondientes EC, cuyos datos han sido seguidos de manera natural por los profesionales. Los biosimilares no siguen este proceso2. Las agencias reguladoras, como la EMA, aplican rigurosas reglas y exigencias a los fabricantes de los biosimilares para establecer garantías suficientes de biosimilitud, pero estas se basan en requerimientos diferentes a los que se exigen para nuevos medicamentos originales. En este sentido, el caso de los biosimilares de los anticuerpos monoclonales, por su complejidad, es especialmente significativo7. En la tabla 1, trasladada desde una publicación previa8, pueden consultarse en síntesis estos requerimientos. Es fácil comprobar en ella que los EC pueden ser, de manera no obligada, solo una fase final de los estudios requeridos para su aprobación, y aun así con un alcance limitado centrado en demostrar equivalencia sin que sea necesario cubrir todo el abanico de indicaciones.
Descripción sintética de las etapas requeridas por la EMA para la aprobación de un biosimilar tipo anticuerpo monoclonal
| Fase | Descripción |
|---|---|
| Estudios no clínicos | |
| Escalón 1. Estudios in vitro | Comparabilidad demostrable con alta sensibilidad |
| Escalón 2. Decisión sobre la necesidad de realizar estudios in vivo | Juicio sobre si los estudios in vivo deben adicionalmente complementar los estudios in vitro |
| Escalón 3. Estudios in vivo | Estudios adicionales en animales que puedan ser necesarios en farmacocinética, farmacodinamia o seguridad |
| Estudios clínicos | |
| Escalón 1. Farmacocinética | Estudios en voluntarios sanos (eventualmente en pacientes si hay criterios de toxicidad) |
| Escalón 1. Farmacodinamia | Requeridos como apoyo adicional a la comparabilidad |
| Escalón 2. Estudios clínicos | Ensayos clínicos de equivalencia de la suficiente potencia, aleatorizados en grupos paralelos preferentemente ciegos |
Adicionalmente a este cambio de paradigma, real al menos en la percepción de los clínicos, los biosimilares han abierto la puerta a un complejo, y a veces interesado debate, de cara a un uso inducido de los mismos en base a decisiones no estrictamente clínicas. Sin duda, y esto debe quedar claro, el clínico tiene la estricta obligación moral de considerar los elementos relacionados con el uso adecuado de los recursos y la eficiencia9. La eficiencia no es un valor optativo. Eficiencia puede entenderse como alcanzar los debidos resultados con calidad empleando los menores recursos posibles. Esto no es ahorrar, es optimizar los recursos disponibles para alcanzar más y más resultados de salud. La eficiencia es la llave de la maximización del beneficio para el conjunto de los pacientes, evitando injustas actuaciones ineficientes en el día a día con pacientes individuales. La eficiencia es un valor que no solo puede, sino que debe, incluirse en el proceso de deliberación clínica. Es decir, la máxima posibilidad de eficiencia se consigue con la suma de actos médicos eficientes; gestionando recursos ajustados a resultados, con efectividad y seguridad objetivas, estableciendo opciones ventajosas desde una perspectiva de coste. Esto es perfectamente asumible en el proceso de consentimiento informado, con una leal explicación al paciente de las razones que apoyan en cada una de las opciones el adecuado equilibrio entre beneficio y coste; el primero manda, pero el segundo no es ajeno, acompaña y participa en la decisión. El problema surge cuando los criterios de coste pretenden ser impuestos desde estructuras de gestión. Claro que los responsables de gestión deben ocuparse de promocionar el uso adecuado de los recursos. Pero lo que está fuera de lugar es ordenar ser eficientes, lo que es un contrasentido porque la eficiencia clínica es un valor que se obtiene por convicción, no por imposición. Un modelo sanitario eficiente lo es, ciertamente, porque sus procesos son eficientes, y dentro de ellos especialmente las decisiones clínicas en los pacientes individuales.
En conclusión, es preocupante que el debate del uso adecuado de los biosimilares esté en ambientes de gestión y no clínicos. Para revertir esto es muy destacable el papel de las sociedades científicas, como SEMERGEN, que al aflorar mediante investigación la atipia que supone el alejamiento de los clínicos de las decisiones de prescripción de los biosimilares basadas en conocimiento clínico, ponen al tiempo en marcha mecanismos de información y formación que permitan rellenar el gap motivado por la ruptura de paradigma que el uso de estos fármacos ha conllevado.
