




INTRODUCCIÓN
La presencia de un cuadro clínico de hipercortisolismo (síndrome de Cushing) plantea un desafío diagnóstico debido a las diversas etiologías de esta entidad (adenomas hipofisarios y adrenales, glucocorticoides exógenos, carcinomas corticosuprarrenales, hiperplasia adrenal, situaciones de pseudocushing, etc.). Asimismo, el hiperandrogenismo en la mujer presenta varias posibilidades diagnósticas (síndrome de ovarios poliquísticos, hipertecosis ovárica, tumores ováricos y adrenales, exposición a fármacos, etc.). Sin embargo, la presentación conjunta de signos y síntomas de hipercortisolismo e hiperandrogenismo, en especial cuando el curso clínico es rápidamente progresivo, eleva la posibilidad de etiología tumoral. Por otra parte, la presencia de hipertensión arterial (HTA) en sujetos jóvenes sugiere la posibilidad de una forma secundaria. Exponemos un caso clínico que ilustra esta situación diagnóstico-terapéutica.
EXPOSICIÓN DEL CASO
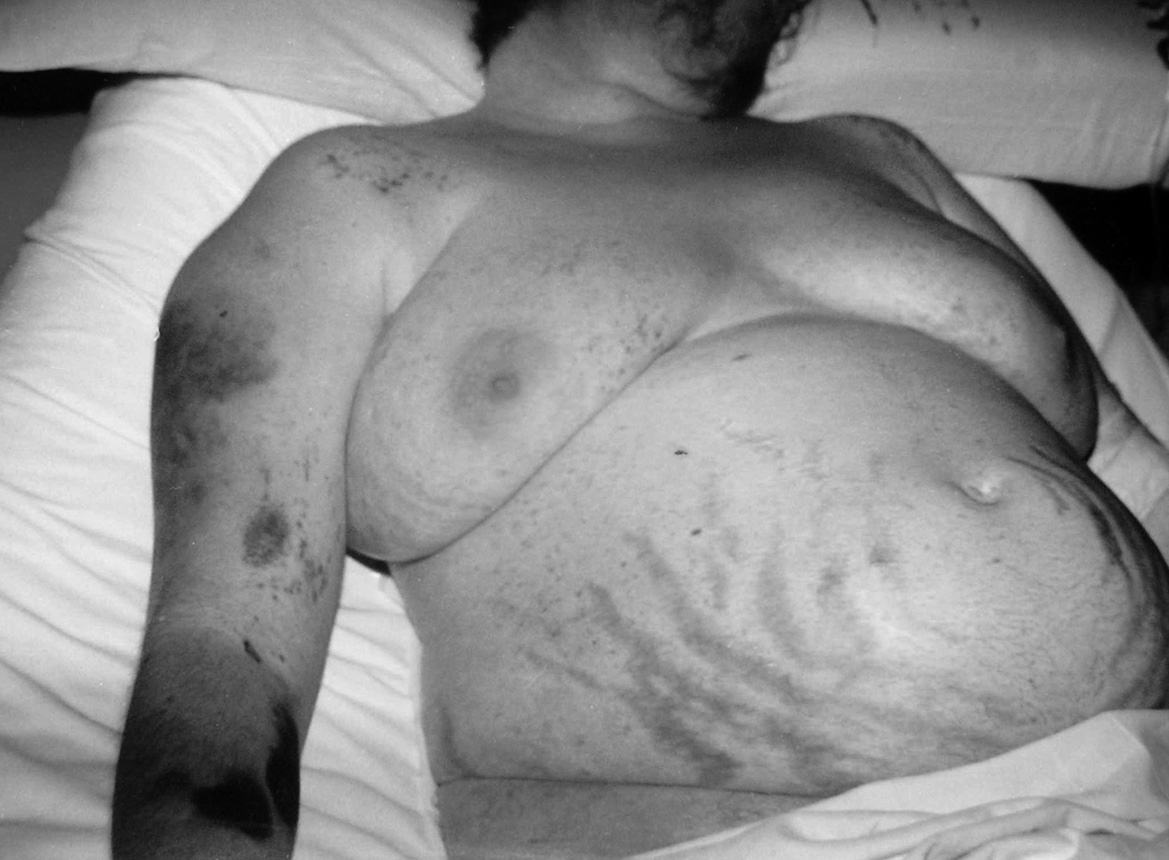
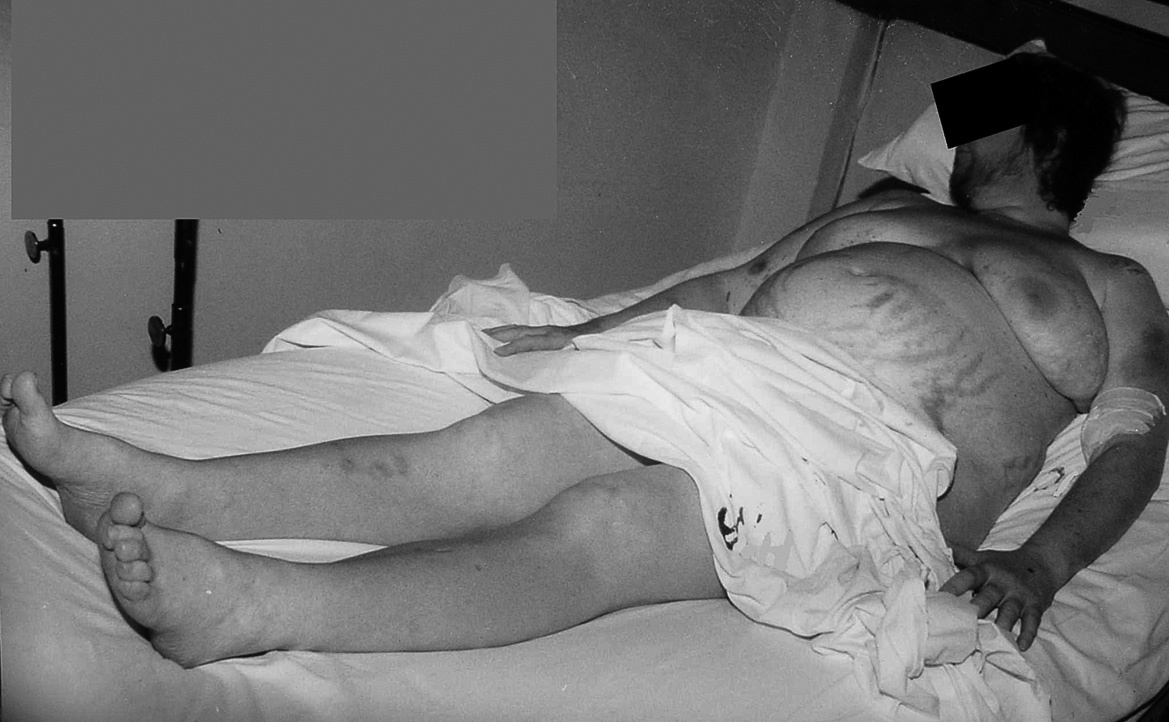
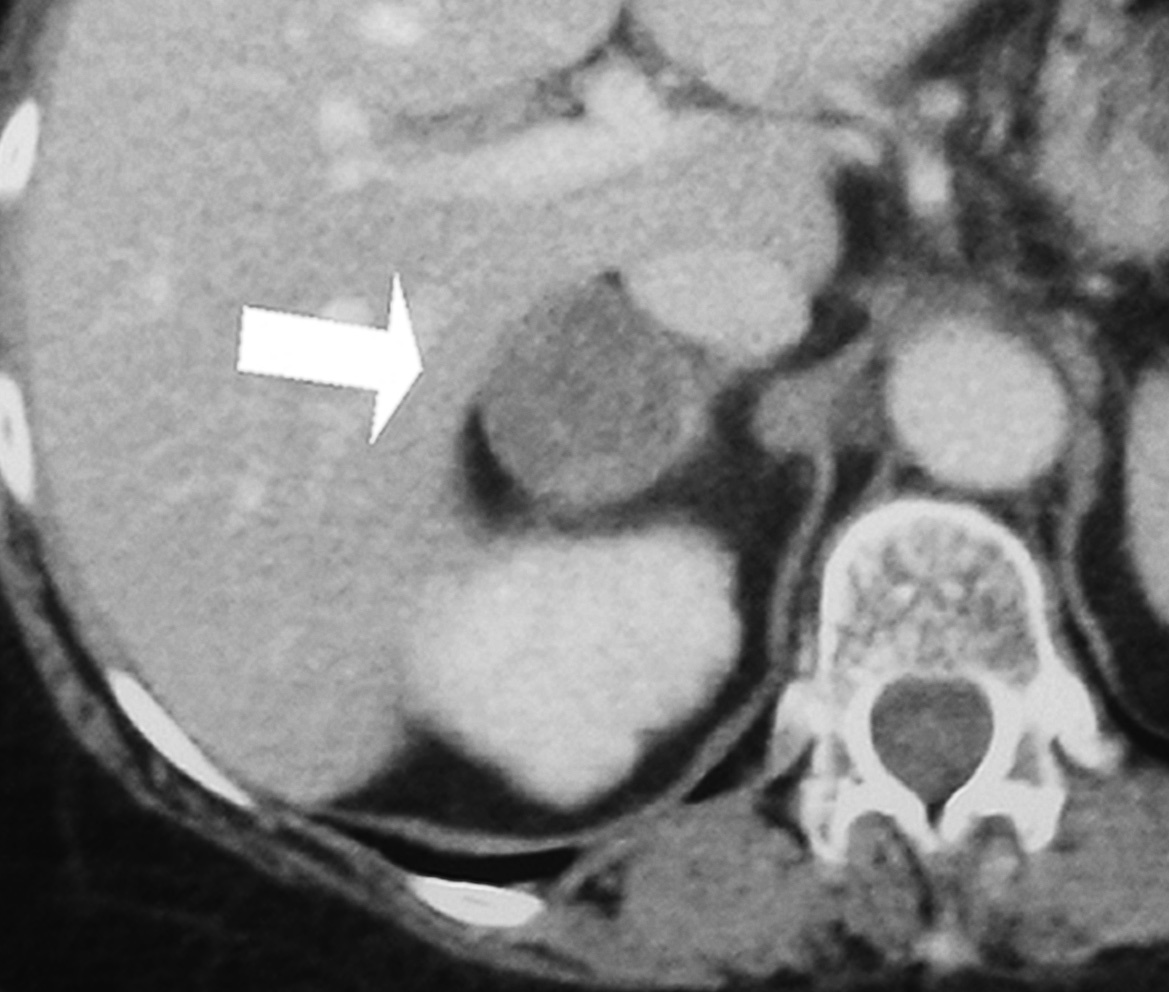
Se trata de una mujer de 34 años sin antecedentes patológicos de interés, que comenzó 8 meses antes del diagnóstico final con HTA (160/90 mmHg), inicialmente controlada con losartán 100 mg diarios, sin evidencias de compromiso de órganos diana. Los valores tensionales se incrementaron progresivamente (hasta 200/100 mmHg), requiriendo el uso de triple terapia antihipertensiva (losartán + hidroclorotiazida + amlodipina). La paciente desarrolló en forma sucesiva un cuadro rápidamente evolutivo caracterizado por incremento ponderal (15 kg en 6 meses), edemas de miembros inferiores y luego generalizados, hirsutismo facial progresivo, debilidad e hipotrofia muscular proximal, ciclos oligomenorreicos y rasgos cushingoides. Fue ingresada en sala de Endocrinología para completar su estudio. Al momento de su admisión destacaba en la exploración física una obesidad de predominio abdominal (índice de masa corporal [IMC]: 33 kg/m2; perímetro de cintura: 104 cm; índice cintura/cadera 0,97), presión arterial (PA) 165/95 mmHg sin cambios posturales, piel fina y lábil con múltiples petequias y hematomas (espontáneos e inducidos por venopunciones previas), estrías abdominales rojo-violáceas "en llamarada" de más de 2 cm de ancho, fascies redondeada, rubicunda, marcado hirsutismo facial con vello terminal abundante y alopecia androgénica moderada (figs. 1 y 2). El examen de genitales externos evidenció hipertrofia de clítoris. No se palparon masas en anexos. Con el diagnóstico clínico de hipercortisolismo e hiperandrogenismo severos con HTA secundaria se realizaron pruebas complementarias cuyos resultados más relevantes fueron: poliglobulia (hematocrito [Hto] 52%; hemoglobina [Hb] 15,5%; eritrocitos 5.150.000/cc), trombocitopenia (80.000/cc); velocidad de sedimentación globular (VSG) 95 mm (1.a hora), hiperglucemia (133 mg/dl); hipoalbuminemia (2,9 g/dl); hipernatremia (Na 149 mEq/l), hipokaliemia (K 3,1 mEq/l); radiografía (Rx) de tórax y electrocardiograma (ECG) sin alteraciones. Pruebas hormonales: hormona tiroestimulante (TSH): 1,4 uµ/ml (0,4-4); prolactina 13 ng/ml (< 20); aldosterona 7 ng/dl (5-20), actividad de renina plasmática 3,2 ng/ml/h (2,9-3,5); adrenalina urinaria 37 µg/24 h (< 50); noradrenalina urinaria 68 µg/24 h (< 90); metanefrinas urinarias 0,8 ng/24 h (< 1,3), 17-OH-progesterona 0,9 µg/l (< 2); hormona luteinizante (LH) 0,5 UI/l (0,8-26), hormona foliculoestimulante (FSH) 1,1 UI/l (1,4-9,6), estradiol 27 pg/ml (20-60); testosterona total 1,7 ng/ml (< 0,9); androstenediona 3,1 ng/ml (< 2,2); sulfato de deshidroepiandrosterona (DHEA-S) 8.300 ng/ml (500-2.500); hormona adrenocorticotropa (ACTH) 8 a.m.: 0,7 pg/ml (10-60); cortisol 8 a.m.: 34 µg/dl (5-25); cortisol libre urinario 245 µg/24 h (20-100); prueba de supresión con dexametasona 8 mg a las 23 h: cortisol 8 a.m.: 21 µg/ml (< 5). Confirmado el diagnóstico de síndrome de Cushing ACTH-independiente e hiperandrogenismo de origen suprarrenal, se realizó tomografía axial computarizada (TAC) toracoabdominal con contraste evidenciando una masa tumoral en glándula suprarrenal derecha, redondeada, heterogénea, de bordes bastante definidos, de 4,5 cm de diámetro mayor, con escasa e irregular captación del contraste (fig. 3). La paciente fue intervenida quirúrgicamente, sin embargo la lesión no pudo ser resecada en forma completa debido a infiltración tumoral de estructuras vecinas. El examen anatomopatológico de la pieza quirúrgica confirmó el diagnóstico de adenocarcinoma suprarrenal primario con invasión extracapsular y metástasis ganglionares paraaórticas (estadio IV). En el postoperatorio persistieron cifras elevadas de cortisol y andrógenos adrenales y se inició tratamiento paliativo con ketokonazol y mitotane (op'-DDD) en altas dosis (2 g/día). La paciente presentó un cuadro de encefalopatía aguda, probablemente inducido por el mitotane, con coma y fallecimiento a los 20 días del postoperatorio.
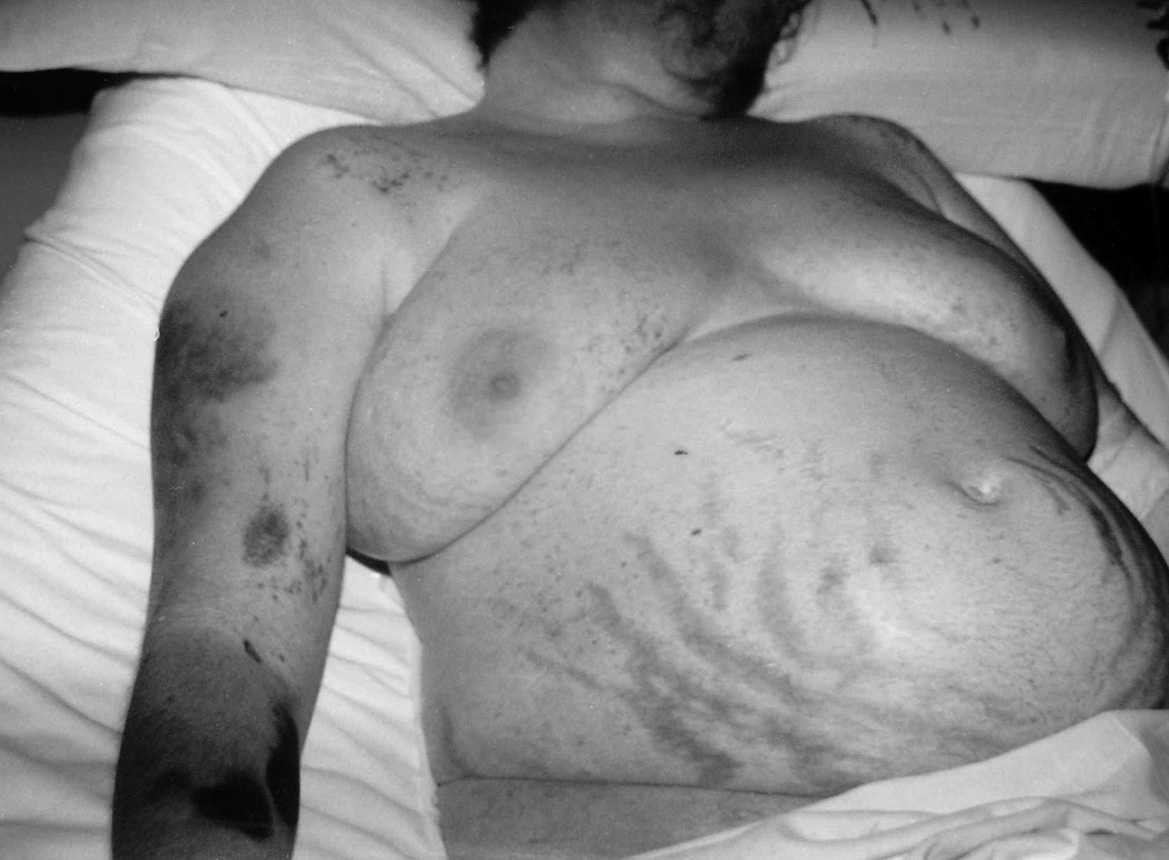
Figura 1. Hirsutismo facial, estrías abdominales, petequias y hematomas cutáneos múltiples.
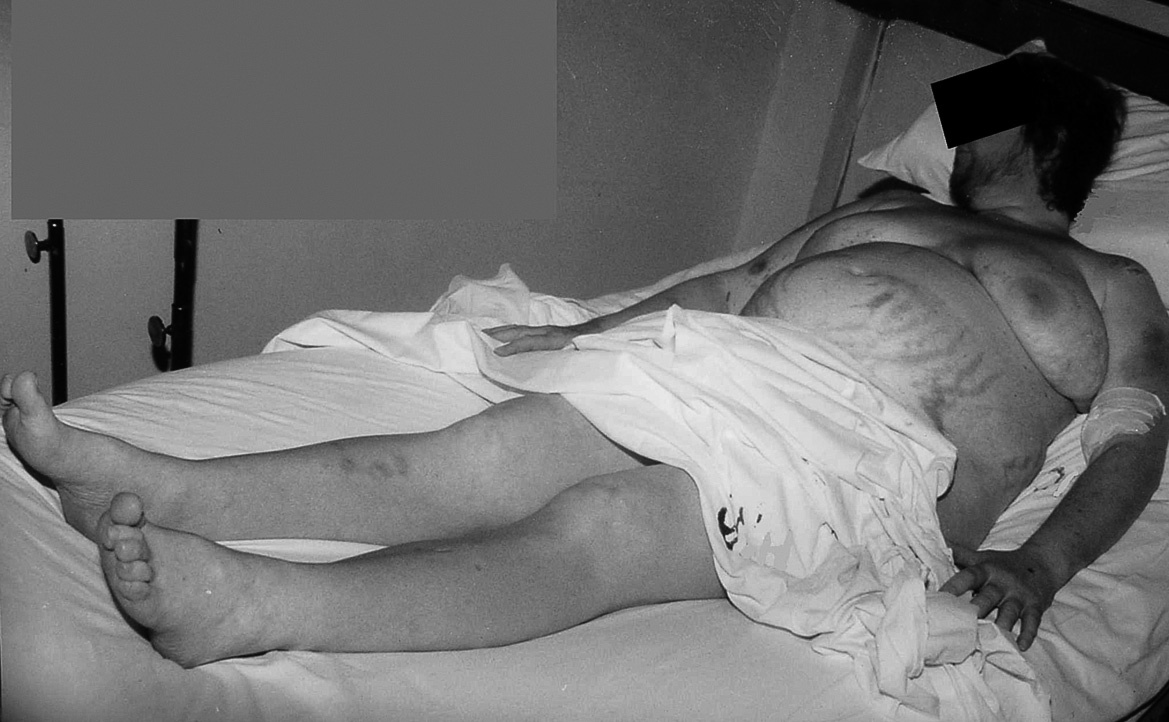
Figura 2. Importante hirsutismo facial y edemas generalizados.
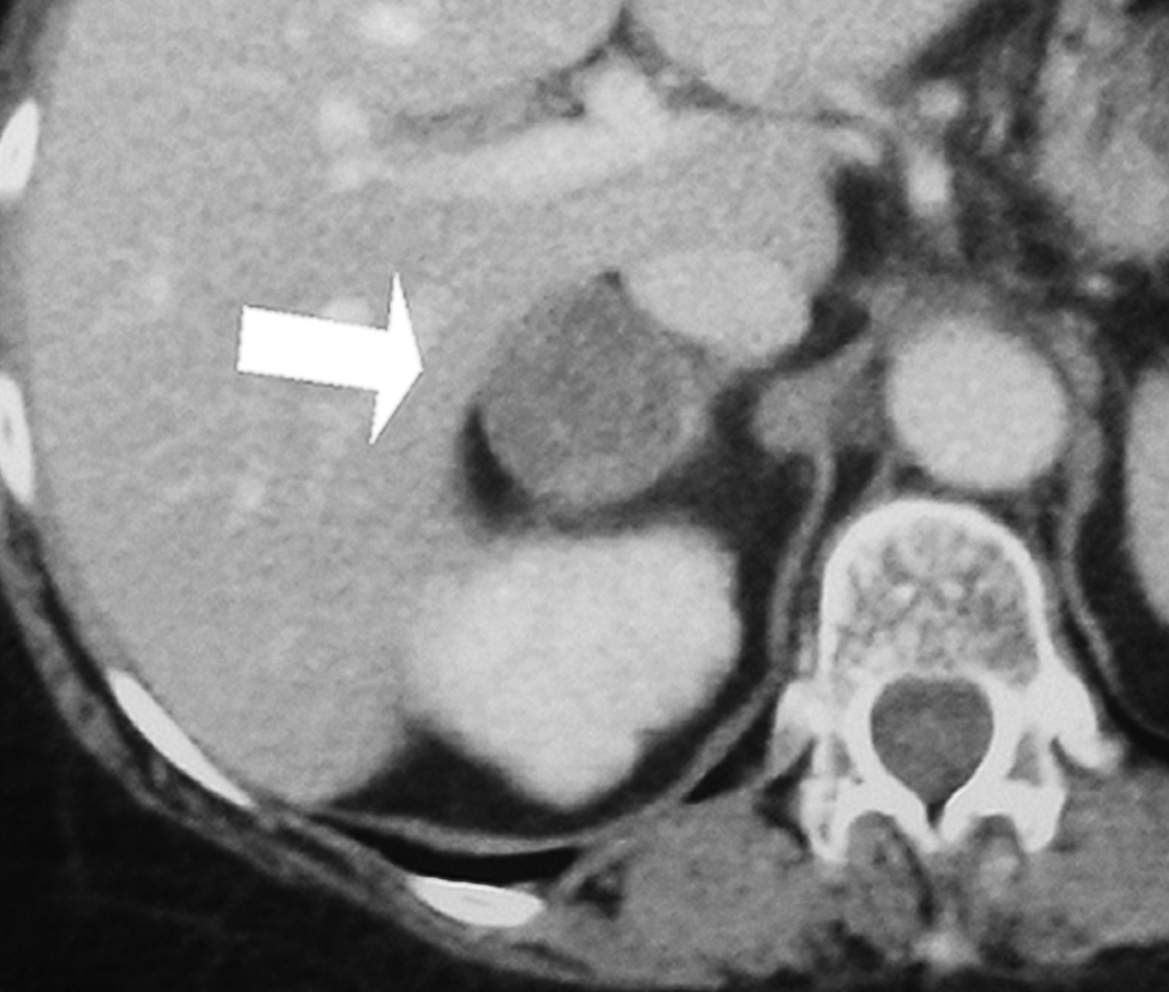
Figura 3. Detalle de la tomografía axial computarizada (TAC) abdominal con contraste IV donde se evidencia una lesión tumoral heterogénea en glándula suprarrenal derecha.
DISCUSIÓN
El carcinoma corticosuprarrenal es una entidad de baja frecuencia, con una incidencia de 0,5-2 casos/millón de habitantes/año1. En cuanto a la edad de presentación, los datos indican un patrón bimodal, con un pico de incidencia antes de los 10 años y otro en la cuarta década de la vida. La distribución por sexos varía entre la series, pero los tumores funcionantes parecen ser más frecuentes en mujeres jóvenes. Existe funcionalidad hormonal en más de la mitad de los casos, principalmente hipercortisolismo (45%) o un patrón mixto, combinado con hiperandrogenismo (45%). Menos frecuente es la situación de hiperandrogenismo aislado (10%) o hiperaldosteronismo (< 1%)2. Es frecuente la HTA secundaria, en cuya génesis intervienen factores hormonales (exceso de glucocorticoides) y ocasionalmente mecánicos (compresión vascular renal). En algunos casos el primer síntoma puede ser el dolor abdominal o raramente una masa abdominal palpable. Conforme evoluciona el cuadro aparecen manifestaciones generales como debilidad, anorexia y pérdida de peso, muchas veces enmascarada por la retención hidrosalina inducida por los glucocorticoides.
El diagnóstico se orienta a partir de la clínica, siendo uno de los datos más orientadores la gravedad y rápida instauración de las manifestaciones por hipersecreción hormonal. Así, un síndrome de Cushing rápidamente evolutivo en pocos meses asociado a hiperandrogenismo severo sugiere la presencia de un tumor corticosuprarrenal. Las evaluaciones hormonales oportunas confirmarán eventualmente la hipersecreción hormonal. El diagnóstico por imágenes se basa en la utilización de la ecografía, TAC o resonancia magnética (RM). Orientan a malignidad la presencia de masas de gran tamaño (> 6 cm), márgenes irregulares y mal definidos, densidad heterogénea, captación irregular del contraste e infiltración de estructuras vecinas3. Se han propuesto 9 criterios anatomopatológicos de malignidad, aunque suele ser evidente por la invasión extracapsular, vascular y/o metástasis (ganglionares, hepáticas)4.
Los tumores suprarrenales se estadifican según la clasificación de Sullivan et al5:
Estadio I: enfermedad limitada a la glándula adrenal, < de 5 cm.
Estadio II: tumor > 5 cm.
Estadio III: invasión local.
Estadio IV: invasión de órganos adyacentes más metástasis ganglionares linfáticas o metástasis a distancia.
La supervivencia global varía entre el 10-20% a los 3 años6, y es particularmente baja en el estadio IV.
Estas características del carcinoma corticosuprarrenal hacen que su tratamiento sea dificultoso y con resultados muy limitados. El paso fundamental es intentar la resección quirúrgica tan amplia como sea posible de la masa tumoral. La posibilidad de exéresis completa es infrecuente debido a que los tumores suelen detectarse en estadios avanzados con extensión extraadrenal. El uso del mitotane (op'-DDD) se plantea fundamentalmente en estadios avanzados o enfermedad no resecable7. Su beneficio en términos de supervivencia no está claramente establecido y su coste y efectos adversos son importantes (náuseas, vómitos, rash cutáneo, diarrea, artralgias, leucopenia, letargo, ataxia, coma)8. Se ha utilizado también en combinación con citostáticos (doxorrubicina, cisplatino, etopósido, vincristina) sin lograr mejores resultados. Existe alguna experiencia con ablación percutánea por radiofrecuencia de tumores localizados.
Así, el caso expuesto ilustra la presentación de un carcinoma corticosuprarrenal en una mujer joven, de un tamaño algo menor del clásicamente descrito, con HTA secundaria, síndrome de Cushing e hiperandrogenismo rápidamente progresivos, diagnosticado en estadio IV y que fallece como consecuencia de la neurotoxicidad inducida por el tratamiento paliativo con mitotane. Asimismo, resalta la necesidad de un alto índice de sospecha clínica para detectar estos casos en estadios más tempranos que eventualmente permitan un mejor pronóstico.
Correspondencia:
G. Alonso.
Avda. Pulianas, 55, 2B. 18013 Granada. España.
Correo electrónico: galonso2@yahoo.com.ar
Recibido el 18-08-05; aceptado para su publicación el 01-02-06.
