






La demencia supone una tragedia personal y una importante carga social, sanitaria y económica. Debido a su actual dimensión, su atención representa un importante gasto sanitario y social, y un elevado coste emocional y económico para el paciente y la familia.
La demencia es un síndrome clínico orgánico, caracterizado por una disminución adquirida, gradual y progresiva de varias de las funciones intelectuales: memoria, orientación temporal y espacial, lenguaje, pensamiento abstracto, capacidad de juicio, etc. sin alteración del nivel de conciencia, y que interfiere de forma significativa en las actividades sociales u ocupacionales del individuo1. Es característico de la demencia un deterioro cognitivo, funcional y los trastornos de comportamiento (agitación, alteraciones de humor, ilusiones, alucinaciones...).
El diagnóstico de demencia es clínico, y se ha establecido el acuerdo de aceptar como válida la especificidad diagnóstica propuesta por los criterios de la Asociación Psiquiátrica2 (DSM-IV) y de la OMS3 (CIE-10) (tablas 1 y 2).


Una vez diagnosticado el síndrome de demencia, el siguiente objetivo es identificar la existencia de algún factor etiológico que pueda ser tratado4. Si bien la mayoría de las demencias son progresivas e irreversibles, el 5-10% del total de las demencias pueden curarse o al menos mejorar notablemente con tratamiento adecuado. En estos casos, es muy importante la identificación precoz para iniciar el tratamiento en la mayor brevedad posible. En el estudio de todas las demencias las pruebas de neuroimagen (tomografía axial computarizada [TAC] y resonancia magnética nuclear [RMN]) son las más apropiadas. Si bien no son concluyentes en el diagnóstico de demencias primarias, su objetivo es descartar causas secundarias como hematoma subdural crónico, hidrocefalia, tumores o lesión isquémica. No obstante, la presencia de atrofia cortical no es patognomónica de demencia. El inconveniente es que no están al alcance del médico de primaria. Las pruebas analíticas descartan más frecuentemente comorbilidad que verdaderas demencias reversibles.
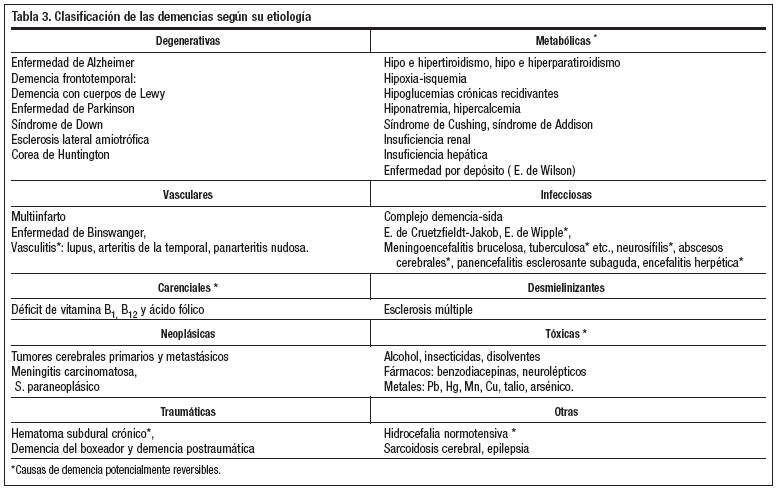
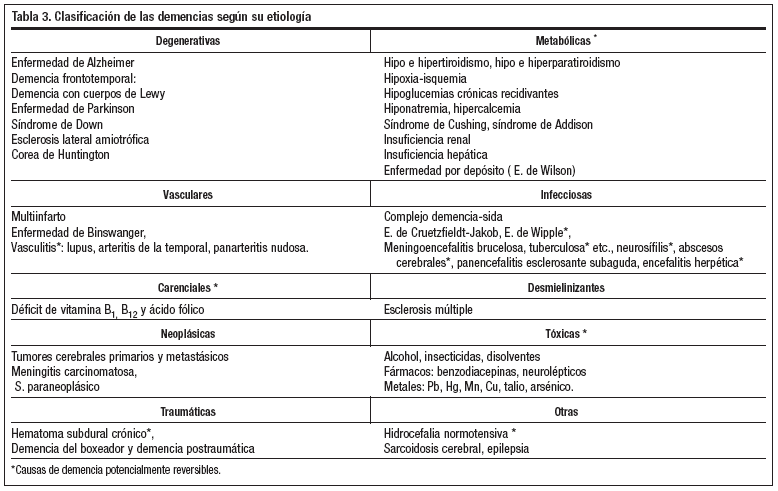
Una posible clasificación de las demencias según la causa se recoge en la tabla 3. Las dos causas más frecuentes de demencia son la enfermedad de Alzheimer (EA) y la demencia vascular (DV), que representan el 80-90% del total de las demencias en el mundo occidental. La EA supone el 55-75% de los casos diagnosticados de demencia en la población geriátrica1, la DV supone el 20-30% y la enfermedad de Pick (EP) tan sólo el 1% de todas ellas.
La demencia frontotemporal es una entidad que incluye a su vez 3 más: a) demencia por EP, b) demencia frontal y c) demencia con enfermedad de motoneurona.
DEMENCIA POR ENFERMEDAD DE PICK
Debe su nombre al profesor de la universidad de Praga, el doctor Arnold Pick (1867-1926), quien en 1892 describió un caso de un paciente de 71 años con deterioro mental progresivo de 3 años de evolución, con trastornos de conducta e importante alteración del lenguaje. Tras su
fallecimiento, encontró en la autopsia una atrofia cere-bral de predominio en el lóbulo frontal y temporal anterior.
Es una demencia degenerativa de tipo cortical, muy poco frecuente y de difícil diagnóstico. La edad de inicio está situada entre los 45 y 60 años, con incidencia similar en ambos sexos5. El comienzo es insidioso, y evoluciona lenta pero progresivamente. La duración media de la enfermedad es de 6-7 años. Cuanto más precoz es el comienzo, más desfavorable es el pronóstico.
CLÍNICA
La enfermedad se caracteriza por cambios precoces y lentamente progresivos del carácter y del comportamiento que evolucionan hacia un deterioro cognoscitivo, con predominio de las alteraciones del lenguaje, y en ocasiones síntomas extrapiramidales (temblor, y alteraciones de la marcha).
Los síntomas más comunes:
1) Alteraciones del comportamiento:
a) Síntomas de desinhibición social: pérdida de cuidado personal, conducta social y sexual inapropiada, y también en relación con el medio ambiente. Irritabilidad, impulsividad adquirida, conductas violentas.
b) Síntomas de inhibición: retraimiento social, indiferencia emocional, pérdida de iniciativa, disminución de interés por las actividades cotidianas y despreocupación sorprendente.
2) Alteraciones cognitivas: (aparecen 1,5-2 años más tarde que las anteriores)6.
a) Lenguaje: empobrecimiento del lenguaje, falta de fluidez, ecolalia, incapacidad para nombrar objetos, disminuye la capacidad para leer y escribir, y finalmente mutismo.
b) Memoria: la pérdida de memoria no es muy llamativa.
c) Orientación, cálculo y gnosias están bastante conservadas. (Gnosias es la capacidad de percibir y reconocer estímulos de los sentidos visuales, corporales y táctiles).
d) Afectación de la capacidad de planificación y ejecución de acciones con un procesamiento mental lento. Dificultad para alternar tareas o realizar actividades motoras secuenciales.
3) Síntomas depresivos:
a) Síntomas de depresión, ideas de suicidio e ideas delirantes. No es raro que comience con sintomatología psiquiátrica5,7.
4) Síntomas neurológicos:
a) Los reflejos de desinhibición cortical aparecen precozmente: reflejo de grasping, orales, de chupeteo, glabelar, palmomentoniano y carneomandibulares.
b) La incontinencia urinaria también es un signo precoz.
c) Síntomas extrapiramidales: temblor, alteración de la marcha, acinesia, rigidez.
DIAGNÓSTICO
Para tener un diagnóstico con certeza es necesaria la biopsia. Las características histopatológicas típicas de esta entidad son la degeneración gliótica, los cuerpos de Pick (cuerpos de inclusión argirófilos dentro del citoplasma neuronal), y las células de Pick (neuronas abalonadas cromatófilas)5,8. La degeneración aparece selectivamente en los lóbulos frontales y parte anterior de los temporales, sin placas neuríticas ni degeneración neurofibrilar en proporción superior a lo que se observa en el envejecimiento normal5.
La analítica de sangre, el líquido cefalorraquídeo, y el electroencefalograma (EEG) son normales.
La TAC y RMN ponen de manifiesto la atrofia fronto-temporal frecuentemente asimétrica, con dilatación de astas frontales. Las técnicas de imagen funcional con TEP (tomografía por emisión de positrones) y con SPECT (tomografía por emisión de fotones) confirman patrones de hipometabolismo fronto-temporal incluso en ausencia de clara atrofia estructural5,6.
TRATAMIENTO
No existe un tratamiento curativo ni tampoco tiene un tratamiento específico.
El pilar básico del tratamiento, como en todas las demencias, son las medidas no farmacológicas de atención al enfermo y al cuidador principal. El médico de Atención Primaria debe proporcionar soporte médico y emocional adecuado al enfermo y a su familia. Es importante que
la familia y los cuidadores conozcan la enfermedad y puedan colaborar favorablemente en la evolución de la enfermedad.
Los síntomas relacionados con trastornos de la conducta parece que mejoran con los inhibidores de la recaptación de serotonina (ISRS)5,8, con neurolépticos y carbamazepina5.
Hay que emplear un tratamiento sintomático para la ansiedad, las alucinaciones, los cambios de personalidad, agitación, insomnio, depresión, etc. Cuando coexisten síntomas neurológicos de tipo extrapiramidal, hay que utilizar los antipsicóticos con cuidado porque pueden agravar el problema.
DEMENCIA FRONTAL
El inicio de la enfermedad se produce alrededor de los 50 años. Aparecen cambios en la personalidad y comportamiento con desinhibición, seguido de trastornos progresivos del lenguaje.
En el estudio histopatológico se aprecia disminución gliótica con espongiosis de lóbulos frontales, pero no aparecen células ni cuerpos de Pick.
DEMENCIA CON ENFERMEDAD DE MOTONEURONA
Presenta la clínica y las características histopatológicas de la demencia frontal y además aparecen fasciculaciones, debilidad y atrofia muscular con parálisis bulbar, debido a
la pérdida de motoneuronas del asta anterior y tronco cerebral.
DIFERENCIAS Y SEMEJANZAS ENTRE EA Y EP
Las diferencias y semejanzas son las siguientes:
1) Las dos provocan demencias de tipo cortical y degenerativa, con mal pronóstico.
2) La EA es muy frecuente (supone el 55-60% del total de las demencias) y la EP muy infrecuente (1%).
3) En ambas el diagnóstico definitivo es anatomopatológico.
4) La alteración de la memoria es menos marcada en la EP que en la EA y tampoco hay apraxia constructiva, síntoma típico de la EA. No hay disfasia sino una reducción progresiva en la iniciativa del lenguaje, que conduce al mutismo. Desde una perspectiva psicológica, en la EA habría un trastorno de las funciones intelectuales, mientras que en la EP se produciría un fracaso en el empleo de estas funciones.
5) Es típico de la EP que comience con trastornos de comportamiento (conducta y personalidad), desinhibición, alteración del lenguaje expresivo con integridad del receptivo.
6) En los estudios de neuroimagen de la EP, la afectación es selectiva de lóbulos frontales y temporales, generalmente asimétricos. En la EA la atrofia cortical es más genera-
lizada, con aumento de surcos, cisuras y dilatación ven-
tricular. Las técnicas de imagen funcional con TEP y con SPECT confirman patrones de hipometabolismo fronto-temporal incluso en ausencia de clara atrofia estructural, a diferencia del patrón temporoparietal de la EA5,6.
En conclusión, si bien la EA supone la mayoría de los casos de demencia diagnosticados en la población geriátrica, hay otras etiologías en las que también debemos pensar, aunque su frecuencia sea escasa.
