




Presentamos el caso de un varón de 36 años sin antecedentes de interés, que tras una micción sufre una pérdida de conciencia y es diagnosticado de síndrome de Brugada tipo I. El síndrome de Brugada es una entidad clínica asociada a síncopes recurrentes que predomina en varones sin cardiopatía estructural. Se trata de una cardiopatía eléctrica que predispone a presentar arritmias ventriculares y muerte súbita. El 70% son esporádicos y el 30% presentan mutaciones en el gen de la subunidad alfa del canal del sodio cardiaco (herencia autosómica dominante). Electrocardiográficamente presenta una elevación del segmento ST en precordiales V1-V3 con punto J elevado que estimula una R’ dando la apariencia de BRD. Se han descrito 3 patrones distintos, demostrándose que el tipo 1 (como el caso que nos ocupa) se asocia con muerte súbita en el seguimiento, por lo que no son necesarios otros estudios complementarios para realizar el diagnóstico.
We present the case of a 36 year-old man with no previous medical history of interest. This man suffered a loss of consciousness before urination and was diagnosed with Brugada Syndrome I. Brugada Syndrome is a clinical diagnosis associated with recurrent syncope. It is predominant in men without structural heart disease. It is a heart disease with risks of electric ventricular arrhythmias and sudden death. The syndrome is sporadic in 70% of patients and a 30% have mutations in the alpha subunit of the sodium channel gene (autosomal dominant inheritance). Electrocardiograms show an elevation in the ST segment from v1 to v3 and in the J-point that stimulates R’, giving the appearance of a right bundle branch block. Three different patterns have been described. Type I (as in this case) is associated with sudden death so no further studies are needed to make the diagnosis.
El síndrome de Brugada es un trastorno eléctrico primario sin cardiopatía estructural descrito en 1992 por Pedro y Josep Brugada1 en el que se asocian síncopes recurrentes y un riesgo elevado de muerte súbita (MS) en adultos jóvenes, y con menor frecuencia en lactantes y niños. Predomina en el sexo masculino2.
Tiene una prevalencia del orden de 5/10.000 habitantes y, excluyendo los accidentes, es la principal causa de muerte de los varones menores de 40 años en las regiones del mundo donde el síndrome es endémico3.
La incidencia real resulta difícil de estimar debido a la existencia de formas ocultas del síndrome, aunque sí se ha detectado una incidencia mayor en los países asiáticos como Tailandia y Japón4.
En los últimos años se han definido los aspectos clínicos, genéticos, celulares, iónicos y moleculares, cambiando con ello los criterios diagnósticos de esta enfermedad.
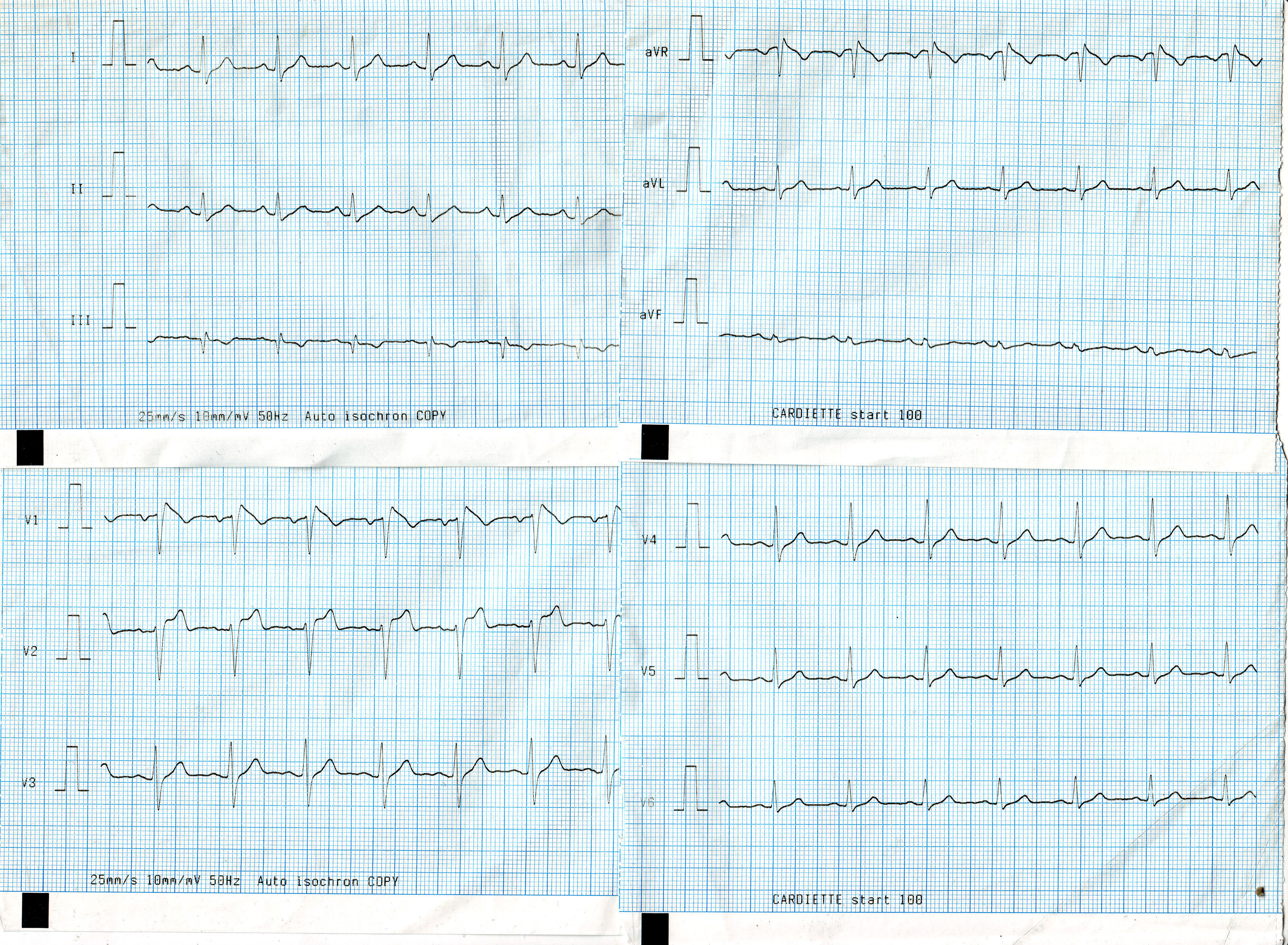
Se trata de pacientes sin cardiopatía estructural, que muestran en el electrocardiograma (ECG) una elevación del segmento ST en precordiales V1-V3 con un punto J elevado que simula una R’ dando la apariencia de bloqueo de rama derecha (BRD)5. No obstante, esta R’ parece ser debida más a una repolarización temprana del epicardio del ventrículo derecho que a un BRD.
Se han descrito 3 patrones de repolarización en precordiales derechas:
- •
Tipo 1: elevación descendente del segmento ST > 2mm en más de una precordial derecha (V1-V3), seguida de ondas T negativas
- •
Tipo 2: elevación del segmento ST > 2mm en precordiales derechas seguidas de ondas T positivas o isobifásicas. (Aspecto de ≪silla de montar≫).
- •
Tipo 3: definido como cualquiera de los 2 anteriores si la elevación del segmento ST es < 1mm.
Los 3 patrones pueden observarse en un mismo paciente, en momentos diferentes, pero también puede presentar un ECG transitoriamente normal.
El patrón electrocardiográfico típico puede ser permanente, transitorio o latente.
El consenso del 2005 unificó diferentes criterios electrocardiográficos, clínicos, familiares, historia de arritmias documentadas o síntomas relacionados, para su diagnóstico.
Son necesarias pruebas complementarias cardiológicas, invasivas o no, que descarten la presencia de cardiopatía estructural.
Se consideran diagnóstico de la enfermedad el patrón tipo 1 (o los patrones tipo 2 y 3 transformados en tipo 1 tras la administración de un bloqueante del canal del sodio), cuando se documenta en combinación con al menos uno de los siguientes criterios clínicos: fibrilación ventricular o taquicardia ventricular polimórfica documentada, síncope, respiración agónica nocturna, inducibilidad de arritmias ventriculares durante el estudio electrofisiológico, historia familiar de MS en edad menor de 45 años o patrón ECG tipo 1 en otros miembros de la familia (documento de consenso publicado en 2002 y 2005)6,7.
Se presenta el caso de un varón de 36 años diagnosticado de síndrome de Brugada tras presentar un episodio sincopal.
Observación clínicaVarón de 36 años que acudió a su centro de salud por síncope autolimitado. En la anamnesis negaba hábitos tóxicos ni alergias conocidas. No explicaba antecedentes patológicos personales ni familiares de interés, incluidos episodios previos de síncope, arritmias cardiacas o muerte súbita. El paciente no tomaba medicación alguna. A las 7:00 h, tras una micción, sufrió una pérdida de conciencia brusca presenciada, sin pródromos, de una duración aproximada de 5 min. En la exploración física se observó un buen aspecto general, una presión arterial de 125/82 mmHg con 93 lat./min, la saturación de oxígeno del 98%, una glucemia de 70mg/dl, temperatura axilar de 36,5°C y una auscultación cardiorrespiratoria normal. El ECG mostraba un eje a 60°, ritmo sinusal, 42 lat./min, una elevación descendente del segmento ST de más de 2mm y onda T negativa de V1 a V3 (fig. 1). No se disponía de ECG previos. Se solicitó una analítica general y de orina urgentes. Se contactó telefónicamente con el servicio de cardiología del hospital de referencia y se programó una visita en 48 h.

El ECG se describió como patrón tipo 1 de Brugada permanente. Se descartaron alteraciones iónicas y cardiopatía estructural por ecocardiograma Doppler.
DiscusiónEl 20% de los supervivientes tras una parada cardiaca por fibrilación ventricular en la que no se asocia cardiopatía estructural esta podría ser debida a un síndrome de Brugada8.
La media de edad de los síntomas en individuos afectados se sitúa entre los 30 y los 40 años, sin embargo, el fenotipo Brugada se puede presentar a cualquier edad.
El 70% de los casos son esporádicos en los que no se ha podido demostrar mutación genética y el 30% restante son de origen familiar con una herencia autosómica dominante9. En estos han quedado demostradas diferentes mutaciones en el gen de la subunidad alfa del canal del sodio cardiaco SCN5A que provocan alteraciones cuantitativas y cualitativas de dichos canales10.
Para la estratificación del riesgo del síndrome de Brugada no es necesario determinar el genotipo. No se ha demostrado que la presencia de la mutación del SCN5A esté asociada a un riesgo alto de paro cardiaco11 por lo que el estudio genético no se realizará de manera sistemática. Únicamente sería útil para el diagnóstico presintomático3. En nuestro caso no se efectuó genotipo ni al paciente ni a sus familiares.
Se han descrito diferentes circunstancias que influyen en la entrada y salida de sodio del canal desenmascarando un síndrome de Brugada oculto como son fiebre, estrés mental, consumo de alcohol, ajmalina, procainamida, disopiramida, propafenona, flecainida, ergotamina, antidepresivos tricíclicos, agonistas alfa-adrenérgicos, antihistamínicos de primera generación (dimenhidrato), cocaína, betabloqueantes, hipopotasemia o disminución del tono vagal12–16. En el caso que nos ocupa, el síndrome de Brugada se desenmascara tras una micción, situación que puede conllevar un estímulo vagal.
La presencia de síntomas es importante de cara al pronóstico. Aunque en su mayoría permanecen asintomáticos, un 17-42% presentan síncope o MS como consecuencia de una arritmia ventricular en algún momento de su vida. Aproximadamente un 23% de los que han sufrido MS ya habían tenido un sincope previamente17. Otros síntomas son convulsiones, trastornos del sueño con respiración agónica y arritmias auriculares18.
La MS cardiaca y las arritmias documentadas aparecen predominantemente durante la noche y en las primeras horas de la mañana19.
El diagnóstico de este síndrome se basa en la historia clínica completa, el ECG y los registros electrocardiográficos seriados. Son necesarias pruebas complementarias cardiológicas, invasivas o no, que descarten la presencia de cardiopatía estructural.
En el caso que presentamos el paciente presentó un ecocardiograma Doppler con parámetros normales.
Actualmente disponemos de estudios que han demostrado que el patrón ECG tipo 1 se asocia con MS en el seguimiento por lo que no son necesarios otros estudios complementarios para realizar el diagnóstico17.
El valor del estudio electrofisiológico es un tema a debate. El estudio electrofisiológico completo se practica a pacientes sintomáticos con ECG con patrón 2 y 3, y a los asintomáticos con ECG basal anómalo, sin embargo, no posee valor diagnóstico en los supervivientes de una fibrilación ventricular con ECG típico11.
Según el Consenso 2005 su indicación en el paciente asintomático con ECG tipo 1 espontáneo es de clase IIa, y en el paciente asintomático con ECG tipo 1 no espontáneo es de clase IIb17.
A nuestro paciente, a pesar de que según lo explicado no sería necesaria la realización de estudio electrofisiológico, se le ofreció esta posibilidad, que rechazó.
En algunos casos complejos puede ser necesaria una prueba farmacológica de inducción mediante la perfusión de bloqueantes del sodio para el diagnóstico diferencial con otras cardiopatías de características similares al síndrome de Brugada como el síndrome de repolarización precoz o la miocardiopatía arritmogénica del ventrículo derecho20.
En cuanto al tratamiento, en la actualidad la única opción terapéutica de eficacia demostrada es la implantación de un desfibrilador automático implantable (DAI)17. Sin embargo, se están buscando otras posibilidades farmacológicas para este síndrome21.
Como conclusión deberíamos incidir en la importancia de tener presente el síndrome de Brugada en las consultas de atención primaria cuando evaluamos a un paciente por síncope, ya que la realización de una anamnesis adecuada, una exploración física meticulosa y un ECG pueden ser suficientes para catalogar a un paciente como ≪paciente en riesgo≫.
Es necesaria una evaluación cuidadosa del ECG y el conocimiento por parte del médico de este síndrome.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.
