




El síndrome de Gitelman es una tubulopatía de herencia autosómica recesiva en el que la alteración fundamental se halla en el túbulo distal, concretamente a nivel del cotransportador Na/Cl, sensible a las tiazidas, codificado en el cromosoma 16q. Cursa con alcalosis metabólica con normotensión, hipopotasemia, así como hipomagnesemia e hipocalciuria que la diferencian del síndrome de Bartter. Su diagnóstico puede demorarse hasta la edad adulta ya que los pacientes pueden mantenerse asintomáticos durante largos períodos de tiempo. El tratamiento consiste en suplementos orales de potasio y magnesio, así como también se ha descrito la utilidad de diuréticos ahorradores de potasio e indometacina.
Gitelman's syndrome is a renal tubule disease of recessive autosomal inheritance in which the fundamental alteration is found in the distal tubule, specifically at the level of the Na/Cl cotransporter, is sensitive to thiazides, and coded in chromosome 16q. It is characterised by a metabolic alkalosis with normal blood pressure, hypokalaemia, as well as hypomagnesaemia and hypocalciuria, which separate it from Bartter's syndrome. Its diagnosis can be delayed up to the adult age, as patients may remain asymptomatic for long periods of time. The treatment consists of oral supplements of potassium and magnesium, and the use of potassium-sparing diuretics and indomethacin has also been described.
El síndrome de Gitelman es una tubulopatía distal de herencia autosómica recesiva. Es un síndrome de curso benigno comparado con el síndrome de Bartter, usualmente diagnosticado en los adultos. La mayoría de los casos son descubiertos casualmente y, a diferencia del síndrome de Bartter, son sujetos hipocalciúricos e hipomagnesémicos. Presentamos el caso de una paciente con hallazgo casual de hipopotasemia secundaria a síndrome de Gitelman.
Descripción del casoMujer de 36 años de edad sin antecedentes familiares de interés. La paciente es diagnosticada de varices y en el estudio preoperatorio para varicectomía se detecta una hipopotasemia de 2,65 mEq/l. Al interrogar a la paciente refiere astenia y calambres en manos y pies de larga evolución (a los que no había dado importancia y por los que nunca había consultado). La exploración física es normal, así como la presión arterial. Se realiza el estudio analítico para diagnóstico diferencial de hipopotasemia con los siguientes resultados: en analítica de sangre destaca: ion potasio, 2,65 mEq/l (3,5-5,5); magnesio, 0,58mmol/l (0,7-1,05); renina (directa) aumentada, 109,6 μU/ml; resto de analítica de sangre normal, incluido el resto de estudio hormonal.
En orina de 24h destaca: calcio, 36,6mg/24h (100-250); fosfato, 1.050mg/24h (300-1.000); sodio, 396mmol/24 h (40-220). Determinación de aldosterona en orina de 24 h dentro de la normalidad. Gasometría venosa: pH, 7,45; pCO2, 46mm Hg; pO2, 37mm; bicarbonato, 32mmol/l; exceso de base, 7mmol/l; SatO2, 74%.
La TC suprarrenal fue normal. Ante la hipopotasemia con alcalosis metabólica, hipomagnesemia e hipocalciuria se realiza el diagnóstico de Gitelman y se inicia tratamiento con suplementos de potasio, magnesio, indometacina y espironolactona. La paciente mejora clínicamente, pero los parámetros analíticos, si bien al inicio mejoraron, posteriormente ha habido que ir aumentando las dosis de los fármacos porque en los últimos controles reaparecía la hipopotasemia. Se realiza estudio familiar siendo las analíticas de los padres normales; los hijos siguen controles en pediatría, aunque no se ha detectado hipopotasemia.
DiscusiónSe describen como tubulopatías un grupo de entidades con manifestaciones clínicas muy diversas cuyo principal denominador común es la alteración en el túbulo renal sin apenas alteración en la función glomerular1. Si bien estas entidades muestran una gran variabilidad en las manifestaciones clínicas, en atención primaria puede establecerse un diagnóstico de sospecha en función de la anamnesis: familiares de enfermedad tubular renal y/o consanguinidad, antecedentes obstétricos y neonatales, historia dietética que investigue especial apetencia por determinados alimentos, ingesta de líquidos y diuresis. En cuanto a la exploración son importantes aspectos como el desarrollo estaturoponderal y el estado de hidratación. La sospecha clínica y una analítica elemental pueden orientarnos al diagnóstico de tubulopatías y en la mayoría de los casos sospechar de qué tubulopatía se trata, aunque en numerosos casos se requiera derivación a un segundo nivel por precisar exámenes complementarios de cierta complejidad (pruebas de función renal, análisis genético, etc.)2.
El síndrome de Gitelman es una tubulopatía distal de herencia autosómica recesiva y se considera que es genéticamente homogéneo dado que la casi totalidad de los casos estudiados son causados por mutaciones diversas del mismo gen3. Este gen, denominado SLC12A3, localizado en 16q13 y formado por 26 exones, codifica el cotransportador del túbulo distal Na/Cl sensible a las tiazidas (también llamado TSC o NCCT). La reabsorción reducida de Na+ en el túbulo contorneado distal ocasiona deficiencia de volumen e hipopotasemia. La pérdida de la actividad del transportador sensible a tiazidas incrementa la reabsorción tubular de calcio, originando la hipocalciuria clásica del síndrome de Gitelman. La hipomagnesemia se encuentra presente en la mayoría de los pacientes con este síndrome y, aunque anteriormente se asumía que estaba relacionada con el defecto en el cotransportador Na/Cl, el mecanismo exacto se desconocía. Recientemente, algunos estudios apuntan a que la pérdida de magnesio podría deberse a la disminución en la expresión del canal de magnesio TRPM6 en el túbulo distal4.
Clínicamente se caracteriza por una gran heterogeneidad, desde pacientes en edad pediátrica que presentan manifestaciones muy claras hasta adultos con síntomas muy leves, incluso casos asintomáticos cuyo diagnóstico se ha realizado a través de estudio genético5.
Los pacientes se encuentran con frecuencia asintomáticos, exceptuando la aparición de episodios recurrentes de debilidad muscular y tetania, que se pueden acompañar de dolor abdominal, vómitos y fiebre. Los intervalos de aparente salud pueden ser muy prolongados y el diagnóstico no suele establecerse hasta la edad adulta. Sin embargo, casi la mitad de los pacientes presentan síntomas menores como apetito por la sal, fatiga, debilidad muscular, dolorimiento general, mareos, nicturia y polidipsia6. El retraso de crecimiento está ausente o es de grado leve. La condrocalcinosis, debida a depósitos de cristales de pirofosfato cálcico deshidratado, es una complicación relevante en adultos7, y también se han descrito artritis de carpo8. En ocasiones son los hallazgos bioquímicos los que nos llevan al estudio y al diagnóstico, de ellos, los más relevantes son hipopotasemia, hipomagnesemia, alcalosis metabólica e hipocalciuria.
En cuanto al diagnóstico de síndrome de Gitelman, se han establecido recientemente unos criterios diagnósticos9: a) hipomagnesenia de origen renal (Mg<1,6mg/dl, en presencia de magnesuria inapropiadamente elevada, EF mg>9%); b) hipopotasemia de origen renal (potasemia<3,6 mEq/l, en presencia de una potasuria inapropiadamente elevada, EF>16%), y c) excreción urinaria de calcio<2mg/kg/día (raramente superior a 0,5mg/kg/día), todo ello en sujetos normotensos y en ausencia de ingesta de diuréticos.
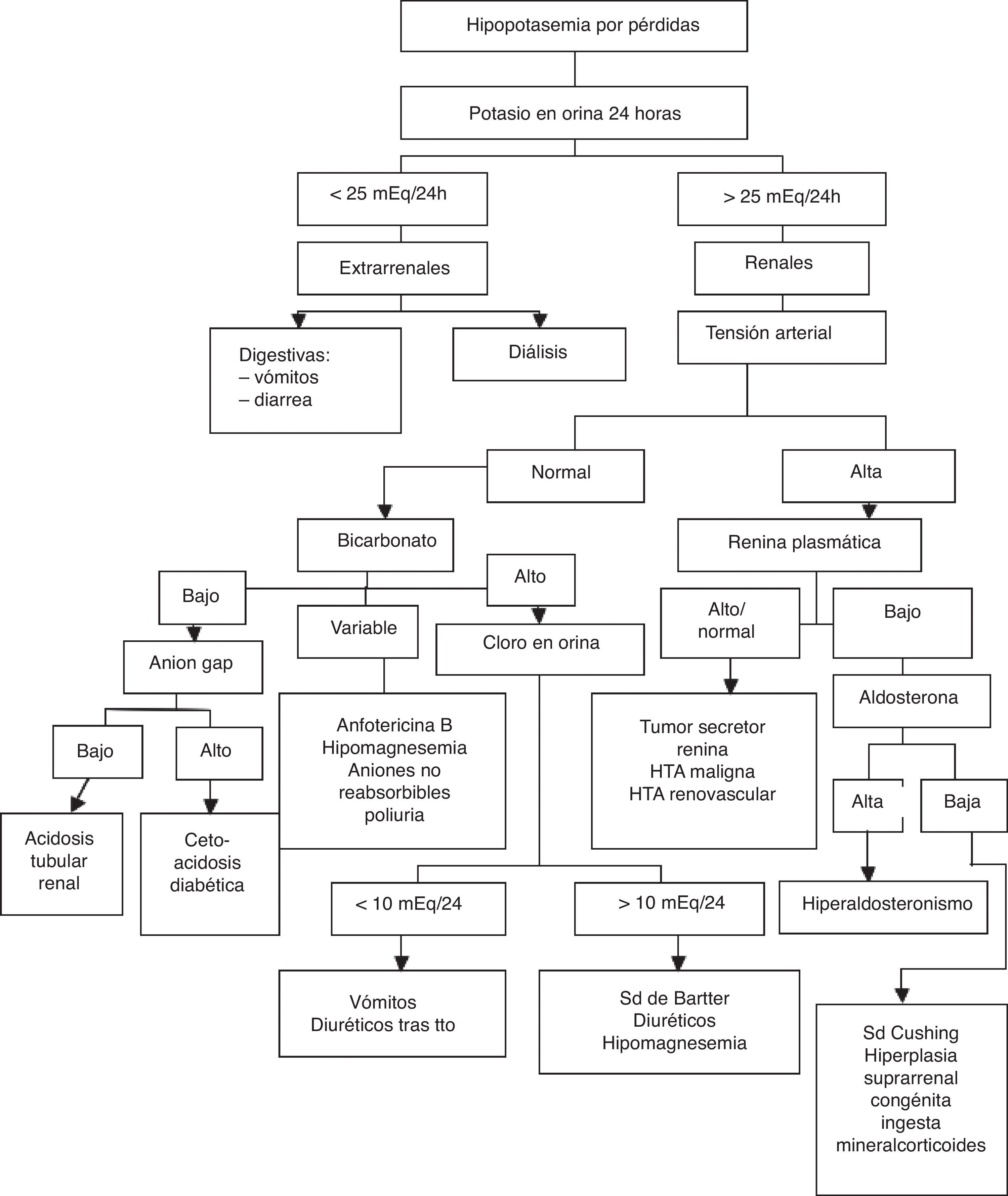
El diagnóstico diferencial se debe realizar en el estudio de las causas de hipopotasemia:
- 1.
Por falta de aporte: anorexia nerviosa, perfusión de líquidos sin potasio a pacientes en ayunas y alcoholismo10.
- 2.
Por pérdidas de potasio (fig. 1).

El tratamiento consiste en la administración de suplementos orales de magnesio y de potasio, reservando la indometacina o los diuréticos ahorradores de potasio para los casos más resistentes11.
Responsabilidades éticasProtección de personas y animalesLos autores declaran que para esta investigación no se han realizado experimentos en seres humanos ni en animales.
Confidencialidad de los datosLos autores declaran que han seguido los protocolos de su centro de trabajo sobre la publicación de datos de pacientes y que todos los pacientes incluidos en el estudio han recibido información suficiente y han dado su consentimiento informado por escrito para participar en dicho estudio.
Derecho a la privacidad y consentimiento informadoLos autores han obtenido el consentimiento informado de los pacientes y/o sujetos referidos en el artículo. Este documento obra en poder del autor de correspondencia.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.
