




El escleredema de Buschke (EB) o escleredema adultorum es una dermatosis rara que se caracteriza por un engrosamiento y endurecimiento de la piel, que inicialmente afecta a cara y cuello, y progresivamente se va extendiendo hacia el tronco y las extremidades superiores1. Su fisiopatología es desconocida, aunque se ha objetivado un aumento de la actividad de los fibroblastos, produciendo un exceso de mucina y colágeno que se acumulan en la capa reticular de la dermis1,2. A continuación se presentan 3 casos de EB de diferente etiología (en todos ellos se ha contado con el consentimiento de los pacientes y se han seguido los protocolos del centro de trabajo sobre el tratamiento de la información y datos de los pacientes):
- 1.
Un varón de 38 años consultó por un cuadro de rigidez de cuello y hombros de un mes de evolución que no había mejorado a pesar del tratamiento con glucocorticoide oral y diazepam. El paciente refería haber padecido un cuadro catarral los días previos. A la exploración, presentaba un endurecimiento marcado de la piel de la parte superior del tronco y del cuello, donde se apreciaba un ligero eritema asociado (fig. 1A). La biopsia cutánea mostraba un aumento de mucina intersticial entre los haces de colágeno compatible con escleredema. La analítica mostró niveles elevados de antiestreptolisina O (ASLO) (694U/ml), sugestivos del antecedente de infección estreptocócica. Se inició, desde el servicio de dermatología, tratamiento con helioterapia (aplicación directa y localizada de luz solar en la zona a tratar), emolientes y ejercicios de movilidad, sin mejoría, por lo que se inició tratamiento con metotrexato 15mg/subcutáneo/semanal, con mejoría parcial tras 4 meses de tratamiento.
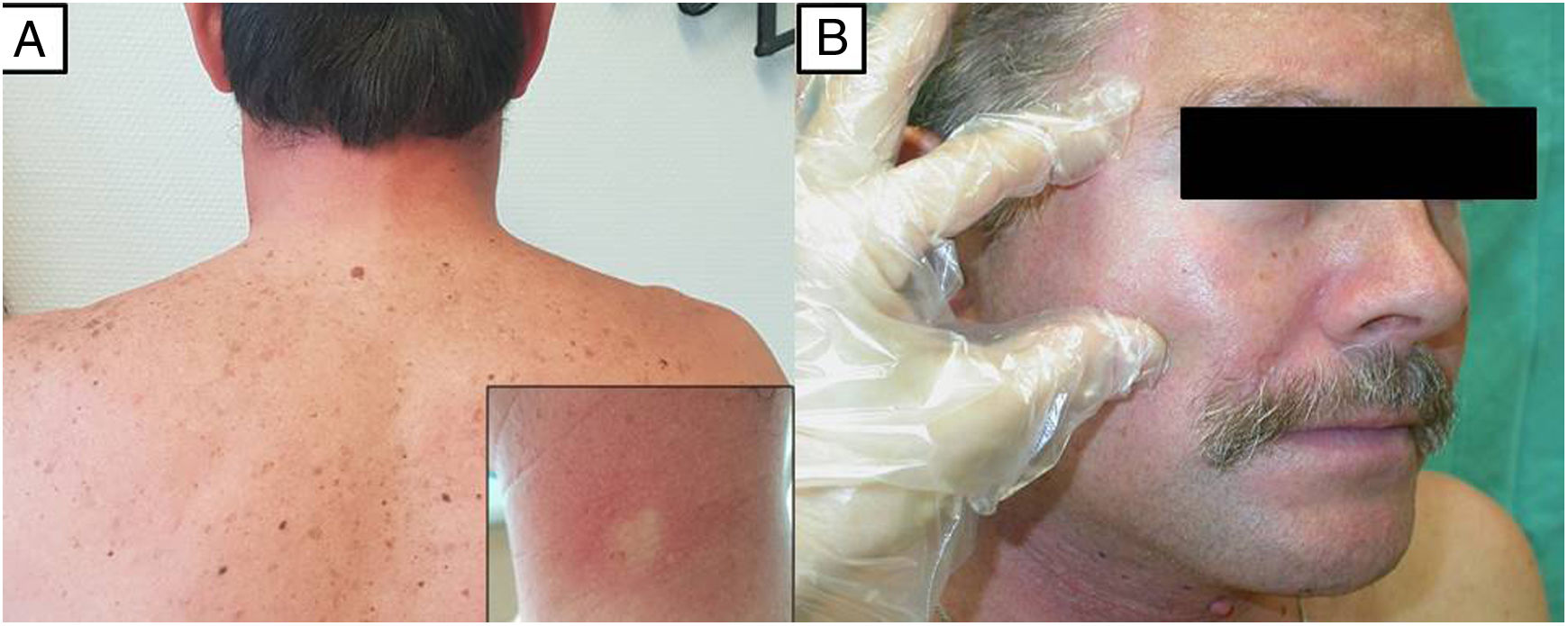
 Imagen clínica del paciente 1, que muestra endurecimiento cutáneo en hombros y región cervical posterior, donde se asocia un leve eritema. Detalle del eritema que blanquea a la digitopresión en la imagen del margen inferior derecho. B) Imagen clínica del paciente 3 que muestra una piel facial endurecida, tensa, con dificultad para ser pellizcada, levemente eritematosa y brillante.") Figura 1.
Figura 1.A) Imagen clínica del paciente 1, que muestra endurecimiento cutáneo en hombros y región cervical posterior, donde se asocia un leve eritema. Detalle del eritema que blanquea a la digitopresión en la imagen del margen inferior derecho. B) Imagen clínica del paciente 3 que muestra una piel facial endurecida, tensa, con dificultad para ser pellizcada, levemente eritematosa y brillante.
(0.14MB). - 2.
Un varón de 49 años refería endurecimiento cutáneo en la región cervical posterior de varias semanas de evolución (fig. 2A). La biopsia fue compatible con escleredema (figs. 2B y C). En la analítica se objetivó elevación del colesterol total (311mg/dl) y de la GGT (58U/l). El paciente reconoció consumo abundante de alcohol, de unos 40g/día, por lo que se le recomendó abandono del hábito enólico y se pautó tratamiento con metilprednisolona aceponato en crema. Dada la escasa mejoría, se inició tratamiento en dermatología con fotoquimioterapia con psoralenos orales y radiación ultravioleta A (PUVA), con leve mejoría.
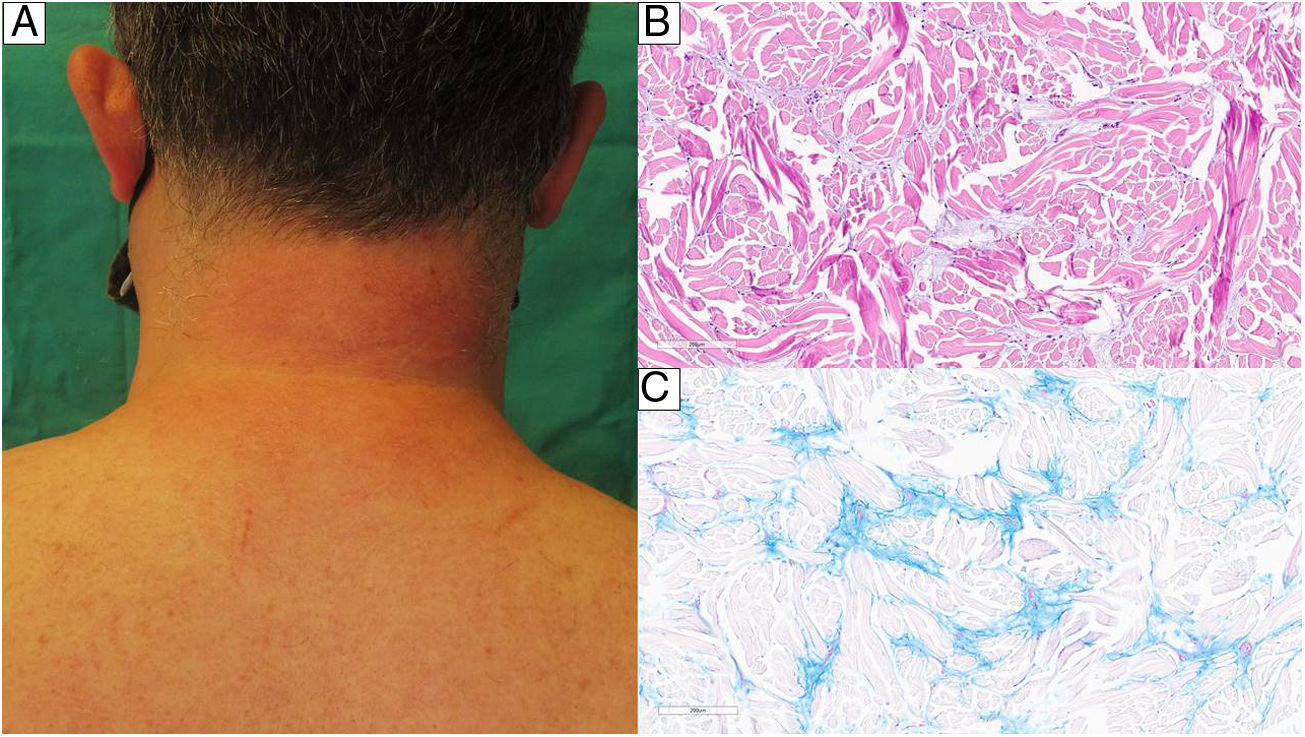
 Imagen clínica del paciente 2 que muestra una piel cervical posterior sutilmente eritematosa y edematosa. B) Biopsia cutánea perteneciente al mismo paciente que muestra haces de colágeno engrosados (color rosa) localizados en la dermis reticular. Entre ellos se observa depósito de un material amorfo, pálido y acelular, que se corresponde con mucina (H&E, ×100). C) Corte histológico de piel teñido con azul alcián (pH 2,5) que tiñe la mucina de color azul (×100).") Figura 2.
Figura 2.A) Imagen clínica del paciente 2 que muestra una piel cervical posterior sutilmente eritematosa y edematosa. B) Biopsia cutánea perteneciente al mismo paciente que muestra haces de colágeno engrosados (color rosa) localizados en la dermis reticular. Entre ellos se observa depósito de un material amorfo, pálido y acelular, que se corresponde con mucina (H&E, ×100). C) Corte histológico de piel teñido con azul alcián (pH 2,5) que tiñe la mucina de color azul (×100).
(0.23MB). - 3.
Un varón de 60 años fue valorado por un endurecimiento cutáneo difuso en la parte superior del tronco, el cuello, la raíz de las extremidades superiores y la cara de 2 años de evolución (fig. 1B). La biopsia fue compatible con escleredema. En la analítica se evidenció un pico monoclonal gamma por cadenas IgG-lambda. No presentaba dolores óseos ni otra sintomatología. Hematología filió el cuadro como gammapatía monoclonal de significado incierto, desestimando el tratamiento. Además, presentó una oftalmoplejia restrictiva por fibrosis orbitaria. Realizó tratamiento por parte de dermatología con glucocorticoides orales, rehabilitación, fototerapia PUVA y UVB de banda estrecha y posteriormente ciclosporina, todo ello sin mejoría, por lo que se decidió tratamiento con inmunoglobulinas intravenosas (IGIV), con leve mejoría.
 Imagen clínica del paciente 1, que muestra endurecimiento cutáneo en hombros y región cervical posterior, donde se asocia un leve eritema. Detalle del eritema que blanquea a la digitopresión en la imagen del margen inferior derecho. B) Imagen clínica del paciente 3 que muestra una piel facial endurecida, tensa, con dificultad para ser pellizcada, levemente eritematosa y brillante.")
 Imagen clínica del paciente 2 que muestra una piel cervical posterior sutilmente eritematosa y edematosa. B) Biopsia cutánea perteneciente al mismo paciente que muestra haces de colágeno engrosados (color rosa) localizados en la dermis reticular. Entre ellos se observa depósito de un material amorfo, pálido y acelular, que se corresponde con mucina (H&E, ×100). C) Corte histológico de piel teñido con azul alcián (pH 2,5) que tiñe la mucina de color azul (×100).")
La prevalencia e incidencia del EB son desconocidas, aunque más del 50% de los casos se presentan en menores de 20 años1,3. Se han descrito 3 subtipos de esta enfermedad en función de su etiología:
- –
Tipo I (55%): afecta a pacientes jóvenes tras una infección respiratoria, principalmente de causa estreptocócica, como en el caso 1. El inicio suele ser agudo y su resolución es espontánea1,3,4.
- –
Tipo II (25%): se presenta en pacientes con una paraproteinemia, como la gammapatía monoclonal o el mieloma múltiple, como en el caso 3. Tiene una evolución crónica con progresión de las lesiones durante años1,3.
- –
Tipo III (20%): también conocido como escleredema diabeticorum, suele afectar a varones a partir de los 40 años con DM tipo 1 y 2 de larga evolución1,3,4.
Existe además, una forma idiopática, como en el caso 2, y se han descrito casos asociados a otras enfermedades como el hiperparatiroidismo primario, la artritis reumatoide, la espondilitis anquilosante, el síndrome de Sjögren, la dermatomiositis, la macroglobulinemia de Waldenström, la púrpura anafilactoide, la cirrosis biliar primaria y el déficit de IgA, además de otros procesos neoplásicos1.
El diagnóstico se basa en la historia clínica, la exploración física y los hallazgos histopatológicos1,4. En la exploración física se encuentra endurecimiento cutáneo en forma de placas «leñosas», localizadas principalmente en el cuello, sin atrofia cutánea. La zona afecta, al ser pellizcada, puede adquirir una apariencia arrugada o en «piel de naranja». Puede presentar una reacción eritematosa transitoria1.
El diagnóstico definitivo se realiza por medio de una biopsia cutanea1,2,5, en la cual se observa un engrosamiento de la dermis, secundario al acúmulo de fibras de colágeno y al depósito de mucina entre ellas1. En el tejido subcutáneo también puede haber una sustitución de la capa lipídica por colágeno de fibras gruesas y característicamente la epidermis se encuentra intacta3.
Las pruebas de laboratorio son útiles para establecer la causa subyacente3. Se pueden encontrar, entre otros, niveles elevados de ASLO, alteraciones del nivel de glucosa y/o la hemoglobina glicosilada (HbA1c), o hiperglobulinemia con elevación de IgG3 en pacientes con gammapatía monoclonal1.
El diagnóstico diferencial debe hacerse principalmente con la esclerodermia y con otras entidades seudoesclerodermiformes como el escleromixedema y la fibrosis sistémica nefrogénica1–3,5. La esclerodermia se caracteriza por endurecimiento de la piel que puede ser difuso, como en la morfea generalizada o en la esclerosis sistémica, o localizado, como en la morfea en placas o en la esclerodermia lineal. La atrofia cutánea, presente en la esclerodermia, es fundamental para hacer el diagnóstico diferencial con el escleredema. Además, también pueden orientarnos al diagnóstico de esclerodermia la presencia de telangiectasias, el síndrome de Raynaud, la calcinosis o las afecciones cuticulares2. El estudio de autoinmunidad sería positivo; e histológicamente, la epidermis estaría atrófica, los haces de colágeno hialinizados y engrosados y habría pérdida de anejos.
El escleromixedema es un tipo de mucinosis primaria que se presenta como placas induradas y una erupción micropapular y que se asocia a gammapatía monoclonal. La fibrosis sistémica nefrogénica se manifiesta como un endurecimiento extenso de la piel en pacientes con insuficiencia renal grave que han estado expuestos a contraste con gadolinio1.
El tratamiento del EB debe ser individualizado, ya que las alternativas terapéuticas tienen escasa efectividad6. Es fundamental detectar y tratar la causa subyacente, lo que puede detener la progresión de la enfermedad y mejorar la sintomatología1, principalmente en el escleredema asociado a paraproteinemia, ya que la afectación cutánea suele ser refractaria. Algunas medidas generales son los baños calientes, los masajes y la fisioterapia6. Además, pueden ser útiles la fototerapia PUVA o UVA2,3,6, la radioterapia con haz de electrones6, la ciclosporina, el metotrexato, la colchicina7, los glucocorticoides tópicos, sistémicos o intralesionales1,6 o las IGIV6. La utilidad de algunos de estos agentes radica en su capacidad de inducir la apoptosis de los fibroblastos dérmicos anómalos o interferir en la señalización de las células, disminuyendo así la producción de colágeno y mucina8.
En conclusión, el EB es una enfermedad poco frecuente, pero cuya clínica es característica y fácil de detectar desde atención primaria. La presentación de estos 3 casos resulta muy útil para conocer las enfermedades sistémicas más frecuentemente asociadas a esta dermatosis, como son las infecciones, la diabetes o la paraproteinemia; así como las claves diagnósticas para diferenciarla de la esclerodermia, evitando innecesarios estudios de autoinmunidad; y realizar un tratamiento individualizado, combinando medidas generales con un tratamiento etiopatogénico.
