




Presentamos el caso de una paciente de 88 años de edad que ingresó en nuestra unidad para estudio de síndrome constitucional de unos cuatro meses de evolución, al que se añadió fiebre elevada y lesiones maculopapulosas generalizadas unos días antes del ingreso. El hemograma inicial mostró una pancitopenia. El diagnóstico sólo pudo realizarse en el estudio post mórtem, siendo éste el de síndrome linfohistiocitosis generalizada y síndrome hemofagocítico asociado a linfoma periférico de las células T.
We report the case of an 88-year-old patient who was admitted in our unit for a study of an appropriate 4-month evolution of constitutional syndrome with high fever, generalized maculopapular exanthema a few days before admission. The initial blood work showed pancytopenia. The diagnosis could only be made in the autopsy study. It was hemophagocytic lymphohistiocytosis and hemophagocytic syndrome associated with peripheral T-cell lymphoma.
La linfohistiocitosis hemofagocítica es una entidad clínica muy poco frecuente, sobre todo en la edad adulta; siendo además su diagnóstico muy difícil, dado que sus manifestaciones clínicas son similares a las de muchas otras entidades clínicas. Asimismo, es muy importante su diagnóstico precoz, ya que sin tratamiento la mortalidad es cercana al 100%; siendo imprescindible para este diagnóstico precoz su sospecha clínica, si es posible desde la atención primaria, lo cual es muy difícil si no se piensa en ello, por lo que creemos de utilidad la divulgación de este tipo de casos.
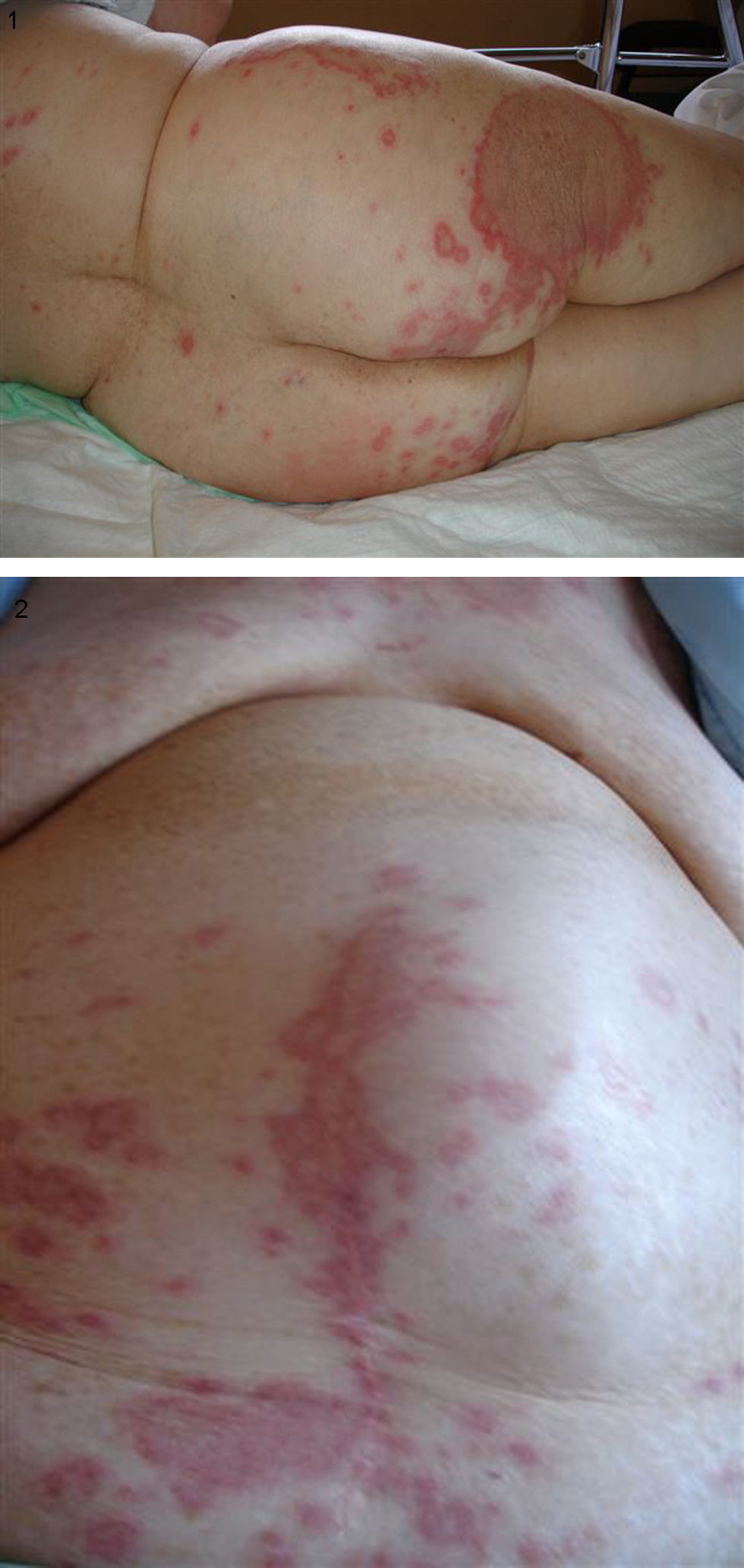
Caso clínicoSe trata de una mujer de 88 años de edad con antecedentes personales de cardiopatía isquemicohipertensiva e insuficiencia renal crónica leve por nefroangioclerosis que consultó por cuadro de unos tres meses de evolución consistente en astenia, adinamia y malestar general progresivo. En los cuatro días previos al ingreso se asoció a lo anterior fiebre de hasta 38°C sin predominio horario ni características bacteriémicas así como aparición de lesiones cutáneas maculopapulosas no pruriginosas ni dolorosas en el tronco, el abdomen y las extremidades (figs. 1 y 2).

A la exploración física, la paciente estaba febril (38°C) y el resto de las constantes estaba dentro de la normalidad, destacando una marcada palidez mucocutánea y un soplo holosistólico (II/VI) en el borde esternal izquierdo y punta, así como lesiones cutáneas eritematosas en el tronco y la raíz de los miembros inferiores de hasta 20cm de diámetro con borde más eritematoso y muchas de ellas con zona de aclaramiento central “en diana” (figs. 1 y 2).
En lo referente a las pruebas complementarias, el hemograma mostró una pancitopenia de 1.000leucocitos/mm3, 7,5g/dl de hemoglobina y 87.000plaquetas/mm3; bioquímica con creatinina de 2mg/dl, AST y ALT (74 y 72U/l), proteína C reactiva (PCR) de 4,24mg/dl y ferritina de 4.520ng/ml. También se realizó determinación de haptoglobina, vitamina B12, ácido fólico, hormonas tiroideas, ANA, ENA, ANCA, factor reumatoideo, hemocultivos y urocultivos seriados y serología frente a Brucella, VHA, VHB, VHC, VIH, CMV y VEB, siendo todas normales o negativas.
Respecto a las pruebas de imagen, tanto la Rx del tórax como un ecocardiograma y una TC toracoabdominal no mostraron alteraciones significativas. Se realizó también una biopsia cutánea de las lesiones antes mencionadas que puso de manifiesto una dermatitis perivascular superficial de predominio linfocitario; asimismo, la biopsia ósea mostró una médula hipocelular con signos de hemofagocitosis moderada e infiltrado intersticial de linfocitos T atípicos.
EvoluciónLa paciente fue tratada inicialmente con transfusiones de hemoderivados así como factor estimulante de colonias (GSF) así antibioterapia de amplio espectro (meropenem, vancomicina y fluconazol) y dexametasona, sin respuesta clínica ni analítica de ésta, la cual fue empeorando de forma progresiva hasta fallecer a las dos semanas del ingreso hospitalario. Posteriormente se realizó el estudio post mórtem, que permitió diagnosticar a la paciente de linfohistiocitosis hemofagocítica y síndrome hemofagocítico maligno asociados a linfoma periférico de las células T (clasificación de la OMS).
DiscusiónLa linfohistiocitosis hemofagocítica (anteriormente histiocitosis maligna) es una entidad clínica de muy difícil diagnóstico, ya que las manifestaciones clínicas ocasionadas por el acúmulo de los linfocitos T activados y los macrófagos en distintos tejidos pueden ser similares a las de otras muchas entidades clínicas.
Este cuadro puede ser primario (si existen alteraciones genéticas) o secundario (si se produce como consecuencia de otros procesos), lo cual crea una importante confusión clínica, no existiendo datos analíticos ni test específicos que permitan diferenciarlos; asimismo, salvo en casos contados, la presentación clínica suele ser idéntica en ambos casos.
Aunque esta entidad es más frecuente en niños desde el nacimiento hasta los 18 meses de vida, hay casos publicados en la literatura médica de aparición en la edad adulta1.
También creemos que es importante resaltar que se puede asociar a distintas enfermedades, entre las que destacan infecciones, que pueden ser virales (citomegalovirus, parvovirus o VEB con mayor frecuencia)2, bacterianas (Brucella y micobacterias, entre otras), por parásitos (leishmania), fúngicas (cándidas), por distintos tipos de inmunodeficiencias, enfermedades autoinmunes (lupus eritematoso sistémico, artritis reumatoidea, sarcoidosis) y secundaria a procesos hematológicos malignos, entre los que destacan las leucemias y los linfomas T, como es nuestro caso3–5.
Como se ha comentado anteriormente, la fisiopatología fundamental es una anormalidad en la disfunción de citoquinas que activan un acúmulo excesivo de los linfocitos T activados y los macrófagos que producen disfunción en todos aquellos órganos en los que se depositan. La clínica inicial de este proceso incluye fundamentalmente fiebre, hepatomegalia, esplenomegalia, síntomas neurológicos, rash cutáneo y linfadenopatía, lo que lo hace indistinguible de otras entidades, por lo que en los casos más agresivos o asociados a enfermedades potencialmente fatales, como es el caso que presentamos, el diagnóstico solamente puede hacerse en estudios post mórtem.
Además, como fenómeno secundario, puede producirse un síndrome hemofagocítico maligno asociado; fundamentalmente cuando coexisten enfermedades neoplásicas (fundamentalmente linfomas de las células T o NK)3,6. Este síndrome es debido a una fagocitosis incontrolada de células rojas y precursores, y aunque suele representar un fenómeno secundario, puede enmascarar la enfermedad de base responsable del cuadro clínico, y está muy relacionada con el pronóstico, debiendo ser las enfermedades neoplásicas la primera sospecha7.
Asimismo, existe controversia en la literatura médica, ya que algunos autores consideran que son dos entidades diferentes y otros como una enfermedad primaria en pacientes con linfohistiocitosis hemofagocítica bien definida mediante criterios clinicopatológicos8. En lo referente al diagnóstico de estas entidades, aunque en la mayor parte de los casos tienen un curso fatal, hay que tener un alto índice de sospecha cuando un paciente presenta varios de los siguientes síntomas: fiebre elevada, rash cutáneo maculopapular, síntomas neurológicos centrales, hepatoesplenomegalia, linfadenopatías, citopenias, coagulopatías, alteraciones de la función hepática y niveles de ferritina.
Aunque, como hemos mencionado con anterioridad, el diagnóstico suele hacerse mediante biopsia ósea, en la que en los casos asociados a neoplasias suele aparecer una médula hipercelular con infiltración de histiocitos y fagocitosis activa7, existen criterios clínicos diagnósticos mayores, debiendo cumplirse al menos dos de los siguientes: fiebre, esplenomegalia, citopenia de al menos dos series, hipertrigliceridemia y/o hipofibrinogenemia y demostración tisular de hemofagocitosis. Los criterios adicionales incluyen una disminución o ausencia de actividad de las células NK, una concentración de ferritina superior a 500μg/l y un receptor soluble CD 25 superior a 2.400U/ml.
En lo que respecta al tratamiento, éste debe iniciarse de forma urgente en el momento que se tenga el diagnóstico de la enfermedad, existiendo el llamado protocolo HLH 94, revisado en 2004 y que está basado en la administración de dexametaxona, etopoxido, ciclosporina y metotrexate, aunque en la actualidad los resultados son desalentadores, estando la mortalidad de este cuadro en torno al 90%9.
