




El síndrome mielodisplásico (SMD) es una enfermedad hematológica, que consiste en una inefectiva hematopoyesis y morfología celular, que produce citopenias y anormalidades en las células sanguíneas1. Constituye, en la población adulta, una de las enfermedades adquiridas más frecuentes, causadas por fallo en el funcionamiento de la médula ósea2. Su diagnóstico puede ser casual tras la realización de un control analítico, aunque puede ser también debido al hallazgo de signos en la exploración física como hematomas3. A propósito de un caso de una mujer que acude a la consulta de atención primaria por un hematoma a nivel de la úvula vamos a realizar una breve revisión de esta enfermedad.
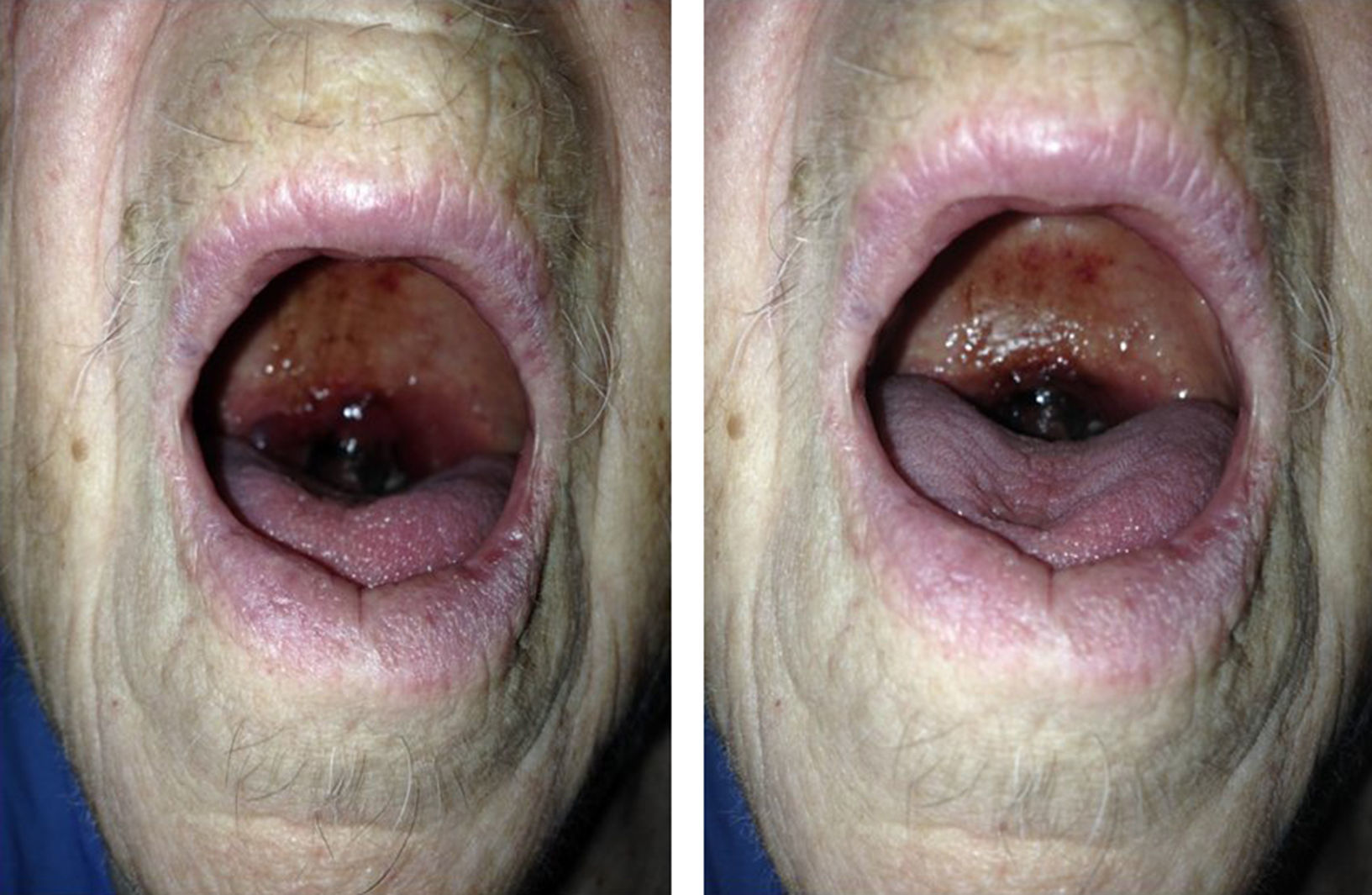
Caso clínicoSe trata de una mujer de 88 años de edad con antecedentes de hipertensión arterial, diabetes mellitus insulinodependiente e hipotiroidismo. No otros antecedentes de interés salvo controles analíticos previos en donde se objetivaba trombocitopenia leve mantenida. Acude a la consulta por cuadro de odinofagia de 2 semanas de evolución y aparición de un hematoma a nivel de la úvula. A la exploración física, la paciente presentaba palidez de piel y mucosas. La auscultación cardiaca y pulmonar no presentaba alteraciones. A nivel abdominal no se apreciaban visceromegalias ni otras alteraciones relevantes. A nivel de extremidades superiores presentaba petequias con hematomas aislados en diferentes fases evolutivas que la paciente atribuida a traumatismos leves repetidos. No se apreciaban adenopatías a nivel cervical. A nivel orofaríngeo destacaba edematización de la úvula con hematoma generalizado con zonas aisladas de necrosis (fig. 1). No se objetivaron alteraciones a nivel del paladar blando y pilares anteriores. No otras alteraciones relevantes.

Ante los signos físicos observados, se realizó un estudio analítico donde no se apreciaron alteraciones bioquímicas. En el hemograma destacaba una discreta anemia macrocítica (hemoglobina de 9,9 g/dl con un volumen corpuscular medio de 103,4fl) con valores de vitamina B12 y ácido fólico normales, sin alteraciones en la serie blanca y una trombocitopenia muy severa (2.000 plaquetas/mm3). Dados los resultados analíticos, se decidió proseguir el estudio de la trombocitopenia de forma hospitalaria en el servicio de hematología del hospital de referencia. En el mismo, se realizó una ecografía abdominal donde no se objetivaron alteraciones relevantes. Se amplió el estudio de laboratorio solicitando un estudio de coagulación y anticoagulante lúpico que resultaron normales. Para el diagnóstico definitivo, se realizó un aspirado de médula ósea donde se confirmó el cuadro de SMD tipo citopenia refractaria con displasia multilinaje (SMD tipo CRDM) dada la displasia trilínea objetivada (más evidente en la serie megacariocítica y granulocítica que la eritroide). El valor de los blastos era inferior al 1%. Se inició tratamiento con corticoterapia para resolver la trombocitopenia, el cual fue refractario, por lo que fue necesaria la transfusión de plaquetas. Tras esto, se decidió alta hospitalaria con tratamiento de soporte consistente en transfusiones periódicas.
DiscusiónEl SMD es una enfermedad frecuente en la población de edad avanzada, aunque su prevalencia es difícil de valorar dado que, en muchas ocasiones, en este subtipo de población se realizan estudios etiológicos de las citopenias observadas en las pruebas analíticas4. En nuestro medio, la edad media de diagnóstico es a los 70 años3, y entre el 85-90% son de causa primaria debidas a alteraciones en la médula ósea relacionadas con la edad. El resto (10-15%) son secundarias a tratamientos tales como ciclofosfamidas o radiaciones ionizantes, las cuales generan alteraciones a nivel genético que favorecen el desarrollo de la enfermedad5. Sea cual sea la etiología del SMD, se caracteriza por tener niveles de blastos en médula ósea inferiores al 20% y la inestabilidad de la enfermedad, la cual puede evolucionar hacia otras enfermedades tales como la leucemia mieloide aguda3.
La alteración analítica más frecuente encontrada en estos pacientes es la anemia (con unos valores medios de 9,5g/dl), la cual suele ser macrocítica (como en nuestro caso) o normocítica6. Otras alteraciones son trombocitopenias y neutropenias. El diagnóstico puede ser casual en un control analítico rutinario por otra enfermedad, aunque el paciente puede presentar sintomatología como fatiga, disnea, escasa tolerancia al ejercicio, infecciones, hematomas o sangrado3. Así mismo, pueden observarse lesiones cutáneas como aftas, adenopatías o exantema fruto de la invasión leucémica7. Una vez sospechado el cuadro, es necesario descartar cuadros como déficits nutricionales (hierro, vitamina B12, ácido fólico, cobre), efectos secundarios medicamentosos (metotrexato, azatioprina), abuso de alcohol, sin infección por el virus de la inmunodeficiencia humana (VIH), o citopenias inmunomediadas que puedan simular el cuadro (lupus eritematoso sistémico, anemia hemolítica autoinmune, etc.)8. El diagnóstico definitivo se realiza mediante el aspirado de médula ósea, ya que nos informa sobre la estructura y variedad celular. La citometría de flujo también es utilizada para el diagnóstico, pero no puede sustituir a aspirado de médula ósea ya que pueden producirse numerosos artefactos durante la técnica9.
El pronóstico de la enfermedad depende de la evolución o no a anemia mieloide aguda, la cual se da entre el 25-30% de las ocasiones. Pese a todo, al tener mayor incidencia en edades avanzadas, el SMD no suele ser la causa de fallecimiento en estos pacientes de forma directa, aunque sí que favorece la evolución de otras enfermedades de base del paciente como las afecciones cardiovasculares10. El cariotipo o las mutaciones genéticas también influyen en el pronóstico y tratamiento, ya que producen aumentos en la sensibilidad a los tratamientos utilizados3. Este se basa en transfusiones periódicas, lo cual es la base del tratamiento de soporte y/o paliativo, o en el uso de factores de crecimiento hematopoyéticos, fármacos inmunosupresores como la lenalidomida, o inhibidores de la metiltransferasa del ADN como la decitabina. Estos tratamientos son indicados en pacientes más jóvenes, con mejor pronóstico y enfocados según las características cariotípicas del SMD. En pacientes en edad infantil se prefiere el alotrasplante de células madre hematopoyéticas, que tiene unas tasas de supervivencia a largo plazo superiores al 50%3.
En conclusión, el SMD es una enfermedad desarrollada principalmente en la población anciana, y que puede cursar asintomática o con sintomatología inespecífica como fatiga o disnea. Anomalías en la exploración física como hematomas pueden estar presentes, que pueden ser atribuidos a otras causas más banales y pueden observarse en localizaciones atípicas, como en nuestro caso a nivel de la úvula. El pronóstico varía según el estadio de la enfermedad y las alteraciones genéticas de base. Es por ello que, una correcta exploración física y atención a las pruebas de laboratorio realizadas, tanto de forma casual o enfocada a esta enfermedad, ha de realizarse en todos los pacientes en los que haya una sospecha diagnóstica de SMD, especialmente en la población joven.
Responsabilidades éticasProtección de personas y animalesLos autores declaran que para esta investigación no se han realizado experimentos en seres humanos ni en animales.
Confidencialidad de los datosLos autores declaran que han seguido los protocolos de su centro de trabajo sobre la publicación de datos de pacientes.
Derecho a la privacidad y consentimiento informadoLos autores declaran que en este artículo no aparecen datos de pacientes.
