




Los encondromas son tumores benignos de cartílago que crecen de forma lenta en las metáfisis de los huesos. Pueden ser lesiones solitarias o múltiples. Las encondromatosis comprenden un grupo heterogéneo de síndromes, difíciles de diferenciar, caracterizados por la presencia de múltiples encondromas que pueden llegar a producir malformaciones musculoesqueléticas (secundarias al acortamiento de extremidades), escoliosis, fracturas patológicas o seudoartrosis. La complicación más temida, el osteocondrosarcoma, puede acontecer hasta en el 25% de los pacientes.
Exponemos el caso de un varón de 67 años, sin diagnósticos previos conocidos, que consulta por la aparición en cadera izquierda de una tumoración dolorosa y rápidamente deformante en el último año. Los antecedentes familiares y los datos clínico-radiológicos confirmaron el diagnóstico de osteocondromatosis múltiple familiar. Aun cuando la evolución clínica y los estudios de imagen hicieron sospechar una degeneración maligna (osteocondrosarcoma), esta no se confirmó en el estudio histopatológico de la pieza quirúrgica.
Enchondromas are benign cartilage tumours that grow slowly in the bone metaphysis. They may involve solitary or multiple lesions. Enchondromatoses include a heterogeneous group of hardly distinguishable syndromes characterised by the presence of multiple enchondromas that may cause musculoskeletal malformations (secondary to limb shortening), scoliosis, pathological fractures, or pseudoarthrosis. The most dreaded complication, osteochondrosarcoma, occurs in up to 25% of patients.
We present the case of a 67-year-old male with no previous diagnosis, requiring attention due to the appearance of a painful tumour in his left hip which degenerated rapidly over the past year. Family history and clinical-radiological data confirmed the diagnosis of Multiple Familial Osteochondromatosis. Although clinical evolution and imaging led to suspect a malignant degeneration (osteochondrosarcoma), this was not confirmed by the histopathological study of the surgical sample.
La encondromatosis familiar múltiple no es una enfermedad frecuente. Aunque suele manifestarse en la primera década de vida la consulta suele ser tardía, bien porque en la familia alguien la presenta y conoce la naturaleza benigna de la enfermedad, que, en muchas ocasiones, no produce molestias al paciente o bien porque, al ser lesiones que se localizan fundamentalmente en las metáfisis, suelen hacerse evidentes y manifestarse clínicamente con cada brote de crecimiento y tienden a estabilizarse al final del desarrollo1.
Los encondromas se localizan principalmente en huesos largos aunque no es infrecuente hallarlos en huesos planos. Las manifestaciones clínicas son variadas, desde casos asintomáticos o simples deformidades estéticas, hasta limitación de la función articular o tendinosa con alteración del crecimiento en los niños o, incluso, grandes deformidades que producen síntomas por compresión de estructuras vecinas2 Aunque generalmente son lesiones benignas, la complicación mas importante es la degeneración maligna a osteocondrosarcoma que siempre se sospechará en caso de crecimiento rápido de una lesión previa y/o aparición de dolor3–5. En estos casos, la cirugía es la piedra angular del tratamiento junto a la quimioterapia pre y post-operatoria.
Caso clínicoUn varón de 67 años de edad consulta por observar en su cadera izquierda la aparición de una deformidad dolorosa y de crecimiento progresivo en el último año.
Jubilado de la construcción, sin antecedentes médico-quirúrgicos de interés, sin hábitos tóxicos ni alergias medicamentosas, no toma tratamientos de forma habitual. No refiere antecedentes familiares importantes salvo que a sus 2 únicos primos hermanos y a una de sus hijas les habían extirpado a los 20 años múltiples «quistes óseos» de etiología no aclarada. La exploración muestra una actitud antiálgica con leve cojera del miembro inferior izquierdo, pérdida de pliegue glúteo contralateral y una deformidad trocantérea izquierda en donde se palpa masa de consistencia pétrea de unos 10cm de diámetro, no dolorosa y sin cambios en la coloración ni en la temperatura.
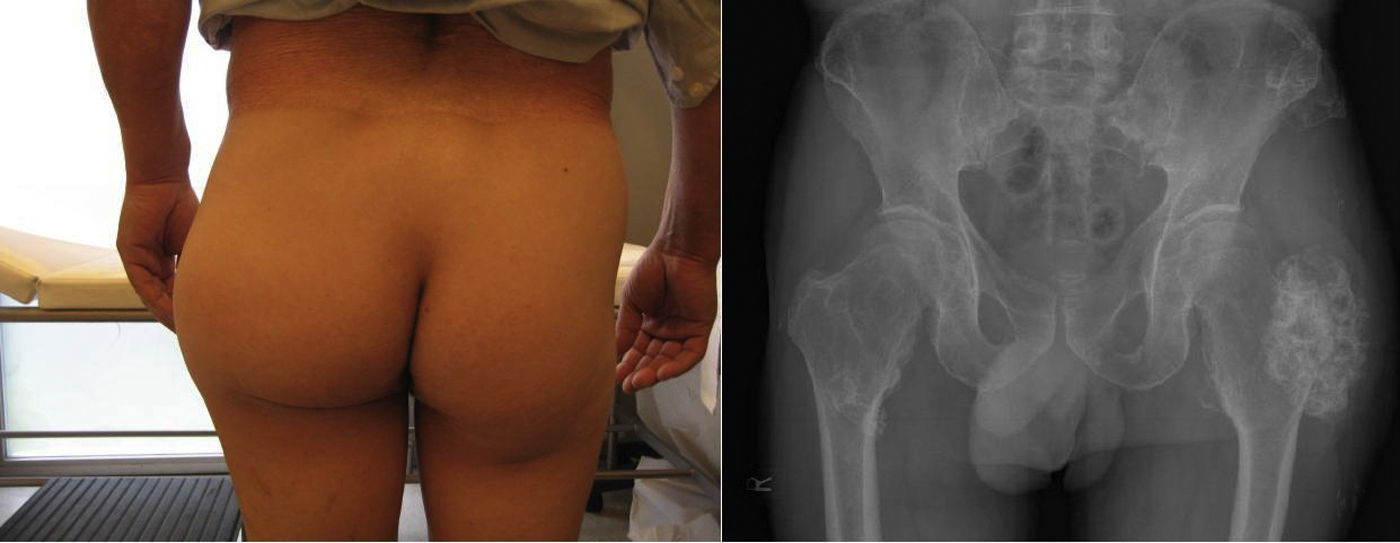
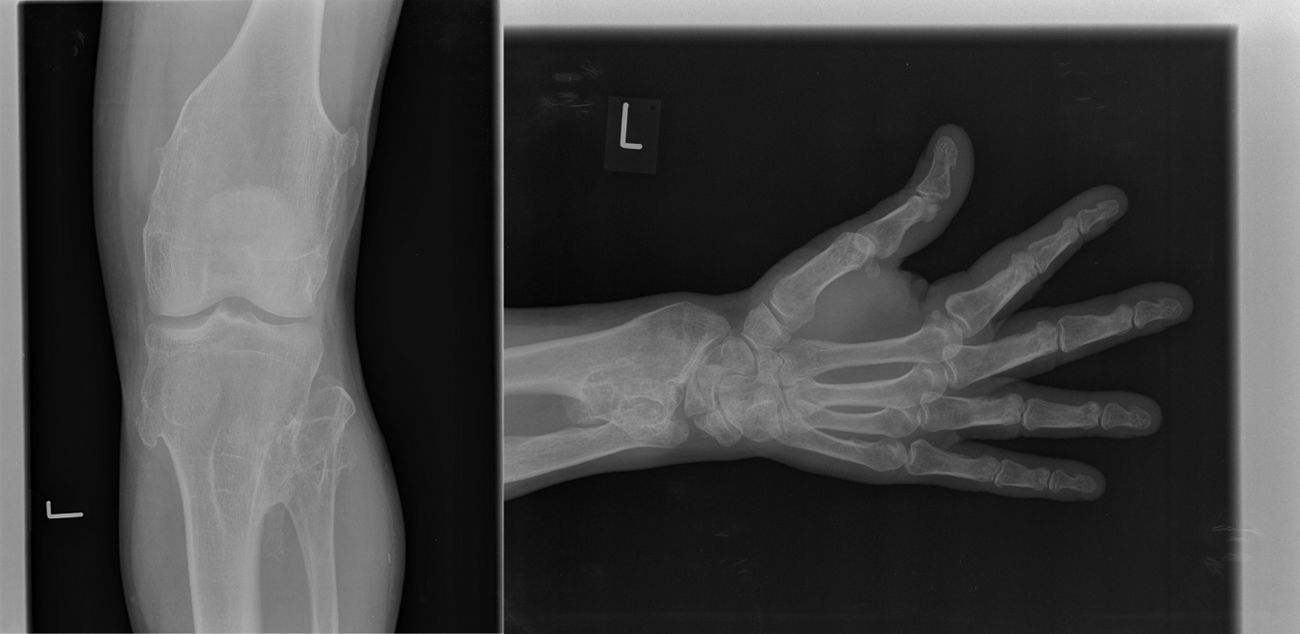
La exploración articular no está limitada funcionalmente y no es dolorosa. En cara dorsal de muñeca y pantorrilla izquierdas se palpan 2 tumoraciones de consistencia dura, fijas y no dolorosas de 2×1 y 1×3cm, respectivamente. La radiología simple muestra múltiples exostosis compatibles con encondromas localizados en: 4.° arco costal derecho, 7.° arco costal izquierdo, extremo proximal de húmero y radio derechos, fémures e ilíacos y extremo proximal de ambos peronés (figs. 1 y 2).
. Deformidad de cadera y proyección antero-posterior de pelvis con encondroma femoral izquierdo gigante y múltiples encondromas en fémur derecho y crestas ilíacas.")
. Osteocondromas en radio, fémur y extremo proximal de peroné.")
Los datos clínicos, radiológicos y antecedentes familiares del paciente orientan al diagnóstico de osteocondromatosis múltiple hereditaria (OMH) pero la aparición de dolor y el rápido crecimiento de la tumoración trocantérea obliga a descartar un probable osteocondrosarcoma secundario. Ante esta sospecha, solicitamos una resonancia magnética (RM) abdominopélvica que muestra una imagen de 99×75×59cm que se origina en trocánter mayor en su cara posterolateral con crecimiento exofítico y parcialmente calcificado, con un grosor del casquete cartilaginoso de 3cm, discontinuidad e irregularidad de la cortical, lo que indicaría probable degeneración maligna de la lesión primaria en osteocondrosarcoma; no hay evidencia de hemangiomas viscerales. El paciente fue intervenido por el servicio de traumatología y se le practicó una resección amplia de la masa trocantérea (tumor y rodete de tejido sano circundante). El posterior estudio anatomopatológico descartó la degeneración maligna de la lesión.
Diagnóstico final: osteocondromatosis multiple hereditaria, osteocondroma gigante en fémur.
DiscusiónLa OMH, aplasia diafisaria, exostosis múltiple cartilaginosa hereditaria o condrodisplasia deformante hereditaria, descrita en 1891 por Bessel-Hagen, es una enfermedad poco frecuente que se hereda con un rasgo genético autosómico dominante con penetrancia variable, aunque se han descrito casos esporádicos. Se han identificado tres loci en relación con la enfermedad: EXT1 brazo largo del cromosoma 8q23-8q24 (responsable del 60-70% de los casos); EXT2 brazo corto del cromosoma 11p11-11p12 (20-30% de los casos) y EXT3 brazo corto del cromosoma 19p6. La OMH se ha descrito asociada al síndrome de Langer-Gienon, la leucemia mieloide aguda, la espondilitis anquilopoyética o el síndrome de Down7.
La prevalencia de la OMH oscila entre 1/20.000-50.000 habitantes, estimándose que en España hay alrededor de 500 familias afectadas. Tiene una clara predilección por el sexo masculino (2:1) y es en este en el que se presentan las manifestaciones clínicas más graves. Los osteocondromas de la OMH se pueden localizar en casi todo el esqueleto adoptando una distribución simétrica y aunque predominan en las metáfisis tanto proximales como distales de huesos largos, no es infrecuente hallarlos en huesos planos1,2.
Los primeros osteocondromas se presentan durante la primera década de la vida y, a diferencia de otras encondromatosis, una vez que se cierran los cartílagos del crecimiento se detiene la formación de nuevos encondromas1,4. Las manifestaciones clínicas son variadas; desde casos asintomáticos o simples deformidades estéticas, a deformidades que producen síntomas por compresión de estructuras vecinas tanto en partes blandas, nervios periféricos o vasos, hasta importantes limitaciones de la función articular o tendinosas que pueden producir alteraciones en el crecimiento de los niños1. Las malformaciones musculoesqueléticas más frecuentes son: acortamiento del cúbito con arqueamiento del radio, asimetría de extremidades, baja estatura, angulación en varo o valgo de la rodilla y deformidades del tobillo4. El diagnóstico de la OMH se establece por la sospecha clínica y se apoya en los estudios de imagen y los antecedentes familiares1,2,4.
La complicación más grave es la degeneración maligna hacia osteocondrosarcoma. Se estima que la transformación sarcomatosa de los encondromas solitarios ocurre en menos del 1%, pero en los pacientes con OMH el riesgo se incrementa hasta el 25% hacia los 40 años de edad2,4,5. Las localizaciones más frecuentes de degeneración maligna son la pelvis, la cadera y la cintura escapular. Siempre se sospechará en los casos de aparición de dolor y/o crecimiento rápido de las tumoraciones1,2,4,5,8 y cuando los hallazgos radiológicos sean característicos: espesor del cartílago superior a 2cm en adultos o 3cm en niños o rotura de la cortical y extensión a partes blandas adyacentes4. En estos casos, la técnica diagnóstica de elección es la RM que ofrece una sensibilidad muy superior a la tomografía computarizada (TC). Es importante resaltar que ni la historia clínica ni la exploración física ni los estudios complementarios bastan por sí solos para determinar el carácter benigno o maligno de una lesión ósea9 por lo que, en los casos de alta sospecha de malignización, se realizará biopsia de aquélla. La biopsia por punción-aspiración mediante aguja fina (PAAF) puede proporcionar resultados falsos negativos, por lo que se recomienda la biopsia escisional, que posee la ventaja de proporcionar suficiente material que disminuya el riesgo de error. Debe completarse el estudio de extensión con TC toracoabdominal y tomografía por emisión de positrones (PET)3,4.
El diagnóstico diferencial se planteará con la displasia epifisaria heminélica, los osteomas, los condromas periósticos, por lo general únicos y, especialmente, con otras encondromatosis múltiples: la enfermedad de Ollier y el síndrome de Mafucci. La existencia de familiares afectados, la bilateralidad de las lesiones, su localización en los huesos largos y el cese de crecimiento de las lesiones al finalizar el desarrollo orientan al diagnóstico de OMH. La localización unilateral, ausencia de familiares afectados y localización más frecuente en manos apoyan el diagnóstico de la enfermedad de Ollier y la unilateralidad y presencia de hemangiomas viscerales y/o mucocutáneos a la enfermedad de Mafucci4,8,10.
La conducta terapéutica está estrechamente relacionada con la sintomatología clínica, variando desde una actitud meramente observacional y conservadora hasta el tratamiento quirúrgico. Este puede abarcar desde la resección del tumor con osteotomías correctivas en los casos más leves, hasta la amputación del miembro afectado en las formas más agresivas e incapacitantes. No existe un tratamiento curativo para la OMH y solo estaría indicado el tratamiento de aquellas exostosis que provocasen deformidades estéticas, trastornos funcionales o dolor y, salvo casos excepcionales, se aconseja posponer la intervención hasta después de finalizar el período del crecimiento con el fin de evitar recidivas1,2,4,7.
En el paciente que presentamos existía una gran sospecha de degeneración a osteocondrosarcoma, tanto por la evolución clínica (dolor y rápido crecimiento) como por las imágenes radiológicas (espesor del casquete superior a 2cm y discontinuidad e irregularidad de la cortical) sorprendentemente el resultado histológico de la pieza descartó degeneración maligna.
ConclusionesLa OMH es una enfermedad hereditaria que se manifiesta en la primera y segunda décadas de la vida, con clara predilección por el sexo masculino. Los encondromas se localizan fundamentalmente en las metáfisis de los huesos largos, crecen mientras el esqueleto es inmaduro y dejan de crecer al finalizar el desarrollo. De carácter benigno, la posibilidad de malignización y el avance de sus deformaciones requiere controles clínicos y radiológicos periódicos con el fin de detectar precozmente sus complicaciones. Se debe aconsejar estudio genético a los familiares de los pacientes afectados.
Responsabilidades ÉticasProtección de personas y animalesLos autores declaran que para esta investigación no se han realizado experimentos en seres humanos ni en animales.
Confidencialidad de los datosLos autores declaran que han seguido los protocolos de su centro de trabajo sobre la publicación de datos de pacientes y que todos los pacientes incluidos en el estudio han recibido información suficiente y han dado su consentimiento informado por escrito para participar en dicho estudio.
Derecho a la privacidad y consentimiento informadoLos autores han obtenido el consentimiento informado de los pacientes y/o sujetos referidos en el artículo. Este documento obra en poder del autor de correspondencia.
