




INTRODUCCION
Dentro de las miopatías inflamatorias, las más conocidas y frecuentes son la polimiositis (PM) y la dermatomiositis (DM). Desde el año 1990 se añade otra entidad, que por sintomatología y características ha merecido ser considerada una entidad propia, es el llamado síndrome antisintetasa (SAS).
El SAS, también llamado síndrome Jo-1, es un trastorno poco definido, considerado como una miopatía inflamatoria idiopática, caracterizado por la presencia de autoanticuerpos antisintetasa en el suero (ACAS). Estos ACAS en verdad son IgG dirigidos contra la enzima sintetasa encargada de unir el ARN con un aminoácido determinado para formar ARN transferente.
Las antisintetasas son el grupo de autoanticuerpos más frecuentes en las miopatías, se han descrito hasta 7 tipos diferentes1, de los cuales el más conocido es el anti Jo-1, que media la unión entre el ARN y la histidina2. Este autoAc es más frecuente en la PM que en DM y se observa en un 50-75% de los casos de SAS, más frecuente en pacientes con HLA DR3. Otros autoAc mucho menos frecuentes son los anti PL-7, anti PL-12, anti EJ, anti OJ, anti DO, recientemente se ha descrito un nuevo autoAc contra la asparraginasa3.
El estudio de autoAc demuestra la presencia de fluorescencia con un patrón citoplasmático característico4,5. Se encuentra asociación entre pacientes con anti Jo-1 y con anti Ro SSA.
Aparte de los AutoAc también encontramos factor reumatoide (FR) en frecuencia creciente, sobre todo en pacientes con afectación articular.
Muchos cuadros de SAS están solapados con PM y DM6, también se asocian en ocasiones con miastenia gravis7, recientemente se han descrito cinco casos secundarios a la toma de hipolipidemiantes orales8.
Los pacientes con SAS presentan una clínica característica con miositis (90%), artritis (50%), fenómeno Raynaud (60%), enfermedad pulmonar inflamatoria (50-70%), fiebre (20%), manos de mecánico (40%), cuadro peculiar consistente en hiperqueratosis, hiperpigmentación y fisuración de las zonas radiales y palmares de los dedos y de las palmas de las manos).
La miositis consiste en una debilidad muscular, dolor y fibrosis, inicio agudo, suele implicar a la musculatura proximal.
El pulmón se ve afectado como una enfermedad intersticial con un amplio espectro de signos y síntomas, siendo la disnea el más frecuente2, tos, hemoptisis e incluso insuficiencia respiratoria aguda. Las pruebas de función pulmonares revelan un patrón restrictivo típico. La radiología torácica suele reflejar un patrón intersticial.
La implicación articular también es muy variada, artralgias, artritis, sinovitis, erosiones óseas, subluxaciones sobre todo en pulgares, síndrome de túnel carpiano.
Otras posibles alteraciones encontradas son la glomerulonefritis proliferativa mesangial6, vasculitis, implicación cardíaca7, calcinosis periarticular8, todas ellas ensombrecen en gran medida el pronóstico, el cual estará determinado sobre todo por el grado de afección pulmonar.
El tratamiento no está estandarizado. Hay pacientes que responden a los glucocorticoides en monoterapia, pero en la mayoría se va a precisar la combinación con inmunosupresores1, incluso durante largo tiempo9.
Los corticoides en dosis de 1 mg/Kg/día junto con azatioprina o metotrexate son utilizados para tratar la miositis, sin grandes resultados.
En caso de afección pulmonar severa se recomienda el uso de ciclofosfamida aplicada en pulsos intravenosos. El tratamiento combinado de corticoides y ciclofosfamida conduce a una mejoría sintomática, regresión de los cambios radiológicos y mejoría de la función pulmonar10.
EXPOSICION DEL CASO
Se trata de una mujer de 50 años de edad, sin antecedentes médico-quirúrgicos de interés salvo intervención de túnel carpiano hace tres años.Hipertensión arterial (HTA) leve en control dietético, en tratamiento con terapia hormonal sustitutiva.
Durante los tres últimos meses acude a la consulta en varias ocasiones por cuadros abigarrados de disnea leve ocasional, febrícula, artralgias difusas, siendo la exploración aparentemente normal y con una buena respuesta ante tratamiento antiinflamatorio y antitérmico convencional.
En el último mes se suma, además, una clínica de debilidad muscular de predominio proximal y un aumento de la disnea a grado II/IV. A la auscultación se observa buena ventilación global, algún roncus aislado sin otros ruidos añadidos. La oximetría da una saturación del 99%. Se solicita analítica destacando únicamente una velocidad de sedimentación glomerular de 45 mm y alteración de enzimas hepáticas (GOT 73 U/l, GPT 195 U/l).
La disnea llega a hacerse de reposo con mal estado general y permaneciendo afebril. Se aprecia a la auscultación una crepitación bibasal con disminución global de la ventilación.
Se solicita una radiografía de tórax donde se observa un infiltrado algodonoso en base derecha, etiquetando el cuadro como neumonía atípica y comenzando tratamiento con macrólidos.
Tres días más tarde refiere comienzo de febrícula y empeoramiento de la disnea, a la auscultación persiste la crepitación, por lo que es derivada a la urgencia hospitalaria donde se la diagnostica neumonía bilateral quedando ingresada en el servicio de Medicina Interna. La exploración y pruebas complementarias se pueden resumir en:
Analítica: 19.000 leucocitos con desviación izquierda, velocidad de sedimentación globular 41 mm, GPT 85 U/l, gammaGT 289 U/l, CPK 351 U/l, LDH 696 U/L, aldolasa 40 U/l. El resto sin hallazgos importantes. Serología para hepatitis B y C negativas.
Exploración neurológica: importante amiotrofia generalizada con debilidad 3/5 de predominio proximal con importante atrofia de los músculos interóseos, lesiones en palma compatibles con "manos de mecánico".
Pruebas reumáticas: anticuerpos antinucleares negativos, AntiENA positivos con especificidad anti Jo-1. C3 y C4 normales, TSH y T4 normales. Índice de protrombina 77%, PTH (parathormona) 48,1 ng/ml.
En el hospital, la evolución es desfavorable, con deterioro clínico y respiratorio, precisando ingreso en UCI por cuadro de distrés respiratorio agudo y fallo renal agudo.
En UCI, aparte de la clínica que motivó su ingreso presenta serias complicaciones, como son sepsis por Staphylococcus coagulasa negativo, shock séptico, diarrea secundaria a Clostridium difficile, polineuropatía del paciente crítico, atrofia muscular y escara sacra.
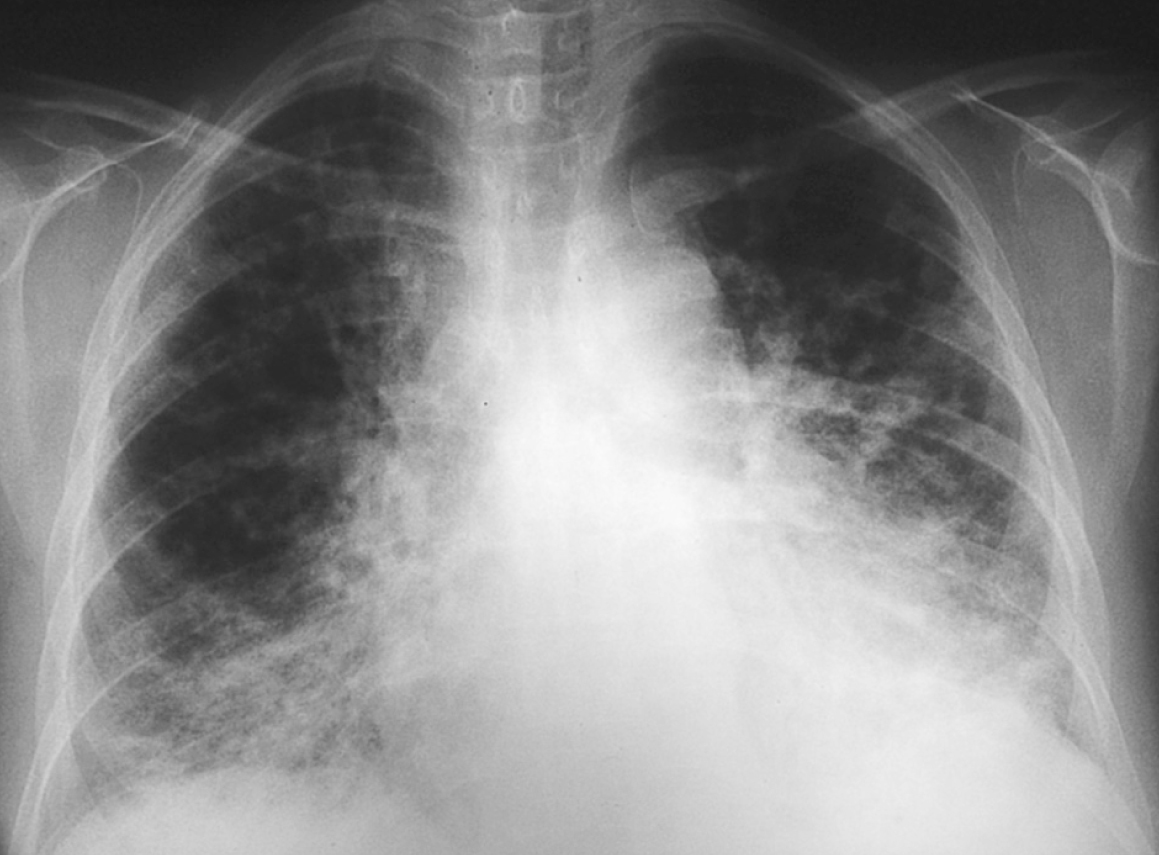
La radiografía de tórax sigue poniendo de manifiesto la persistencia del distrés (fig. 1). Las pruebas de función respiratoria (capacidad vital [CV] 62%, volumen espiratorio máximo en el primer segundo [VEMS] 73,9%, capacidad de difusión pulmonar de CO [TLCO] corregida de 69%) son compatibles con patrón restrictivo leve con afectación del intercambio gaseoso.
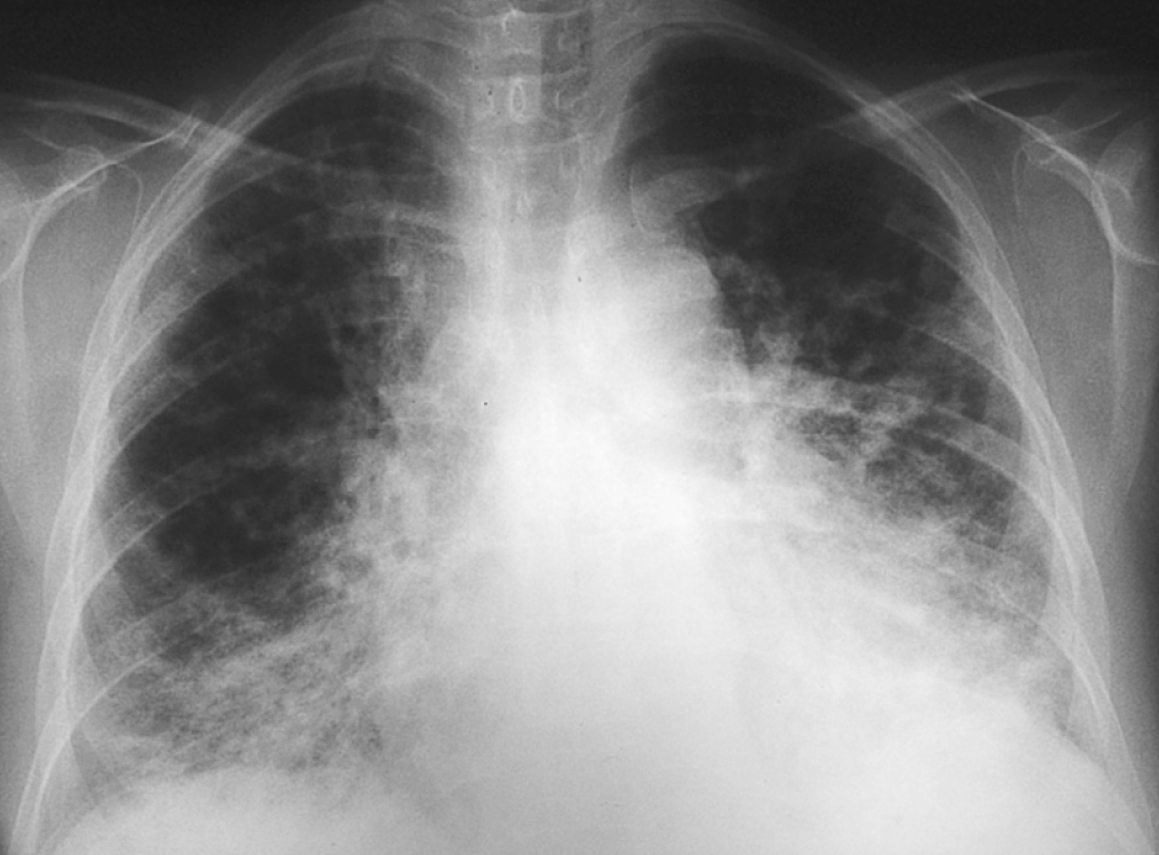
Figura 1. Distrés respiratorio en radiografía de tórax.
Se realiza un ecocardiograma, que denota una fracción de eyección del 70%, con ligera insuficiencia aórtica. La gammagrafía ósea es normal.
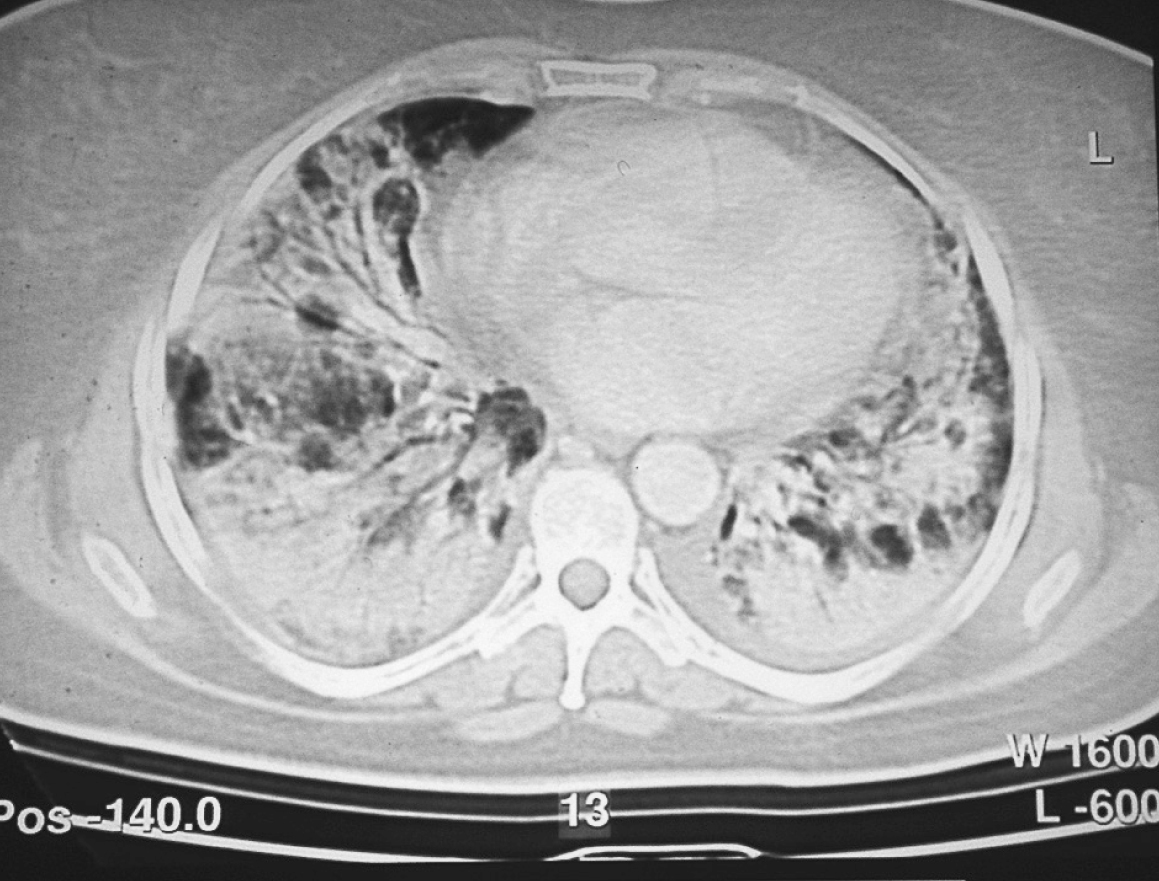
En la tomografía computarizada se aprecia miopatía intersticial con tractos fibrosos y engrosamientos septales en ambas bases pulmonares y patrón en vidrio deslustrado de predominio en vértices (fig. 2).
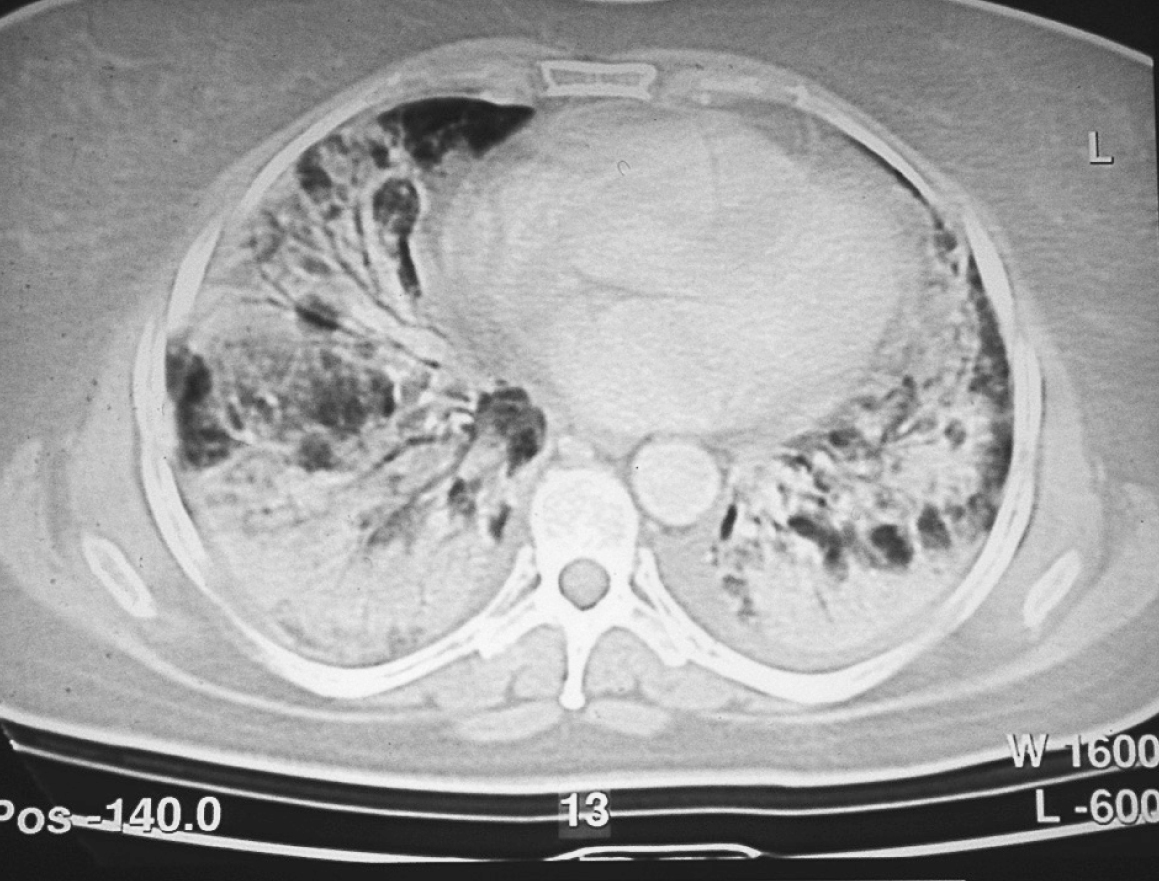
Figura 2. Tomografía axial computarizada: engrosamientos septales. Patrón en vidrio deslustrado.
Presentaba esteatosis hepática difusa.
Tras tratamiento con antibioterapia y dosis altas de corticoides la paciente evoluciona favorablemente, abandonando los cuidados intensivos después de un mes. Se va disminuyendo progresivamente la dosis de corticoides y se le administra un primer pulso de ciclofosfamida, a la vez que se comienza con rehabilitación en el hospital.
En planta, persisten los pulsos de ciclofosfamida y la dosis mínima esteroidea, siendo dada de alta veinte días después.
En domicilio se confirma la mejoría clínica siendo capaz de deambular con autonomía para su alimentación y cuidado. Persiste la debilidad proximal en piernas, con imposibilidad para levantarse de una silla sin apoyo de los brazos. Desde el punto de vista respiratorio persiste un leve grado de disnea I/IV.
DISCUSION
A pesar de que los anticuerpos antisintetasa históricamente se han considerado específicos de la polimiositis y dermatomiositis, desde el año 1990 se define un síndrome propio caracterizado por la presencia de dichos autoanticuerpos junto con una clínica característica de artritis, fiebre, Raynaud, enfermedad pulmonar intersticial y manos de mecánico, cuadro ya conocido como síndrome antisintetasa.
En ocasiones aparece solapada e interrelacionada junto con otras miositis como DM y PM11, pero en otras muchas aparece en solitario o asociado a otras entidades como la miastenia gravis12, incluso se han descrito cinco casos secundarios a tratamiento con hipolipidememiantes orales13.
Existe una gran variabilidad en cuanto a la presentación clínica de este síndrome, descrito en varias series estudiadas por diferentes autores. En nuestro caso llama la atención cómo el espectro de la sintomatología abarca prácticamente la totalidad de las posibilidades: a pesar de haber sido un inicio larvado con fiebre de origen desconocido, la evolución y las complicaciones han sido de una rapidez y envergadura muy considerables.
El estudio radiológico típico del síndrome muestra un patrón intersticial en diferentes estadios evolutivos, como se ha visto, nuestra paciente ha cursado incluso con edema agudo pulmonar.
Las pruebas de función respiratorias típicamente muestran afección intersticial con disminución de la difusión en estadios iniciales, también encontrado en el caso en cuestión.
A la hora de valorar a un paciente con ACAS, es importante constatar que no se sabe la verdadera frecuencia ni el papel que ocupa dentro de otras neumopatías intersticiales para lo cual se haría preciso realizar un seguimiento longitudinal a pacientes con neumopatías.
En cuanto al tratamiento adecuado no hay muchos datos en la bibliografía ni buenos resultados en general. Algunos pacientes mejoran con tratamiento antiinflamatorio o corticoideo, aunque la mayoría van a precisar de tratamiento inmunosupresor durante largo tiempo. En nuestro caso, y a pesar de que la afección pulmonar es la principal predictora de pronóstico y de la cual nuestra paciente tuvo serias complicaciones, la respuesta al tratamiento corticoideo junto a la ciclofosfamida en ciclos puede ser considerada como muy aceptable.
Correspondencia:
C. Fernández Piedra
Avda. Murrieta 24 3.o Izda. 48980 Santurtzi. Vizcaya. España.
Recibido el 18-03-05; aceptado para su publicación el 29-07-05.
