


Inhibidores de la transcriptasa inversa
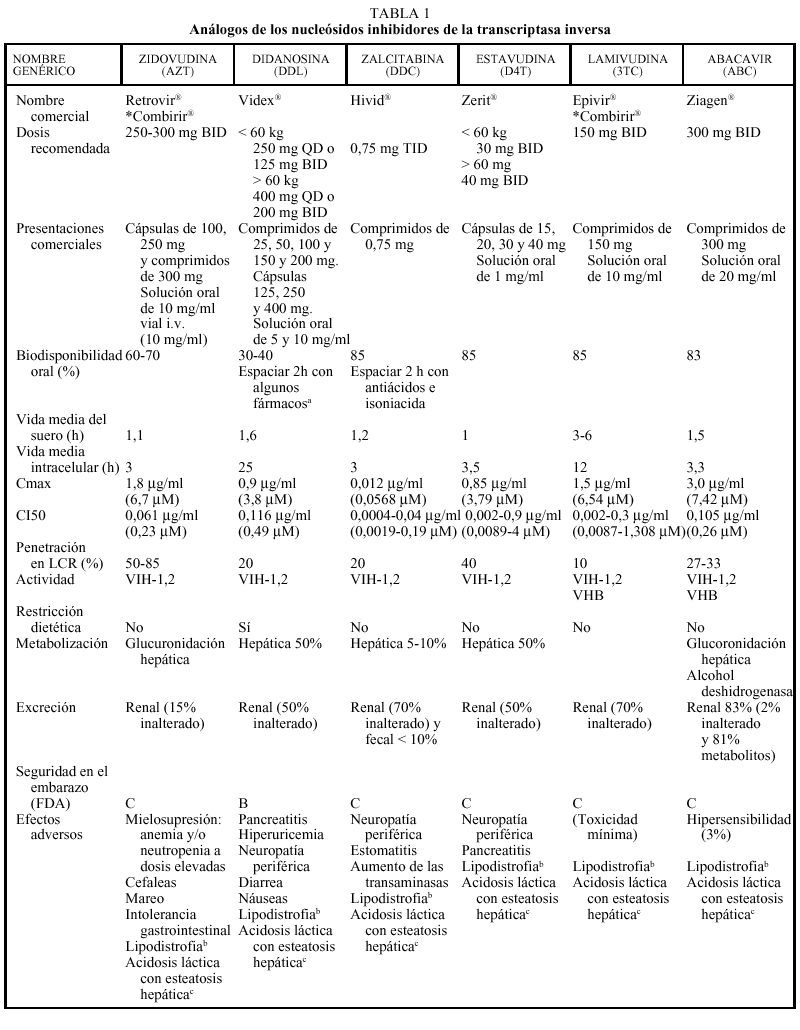
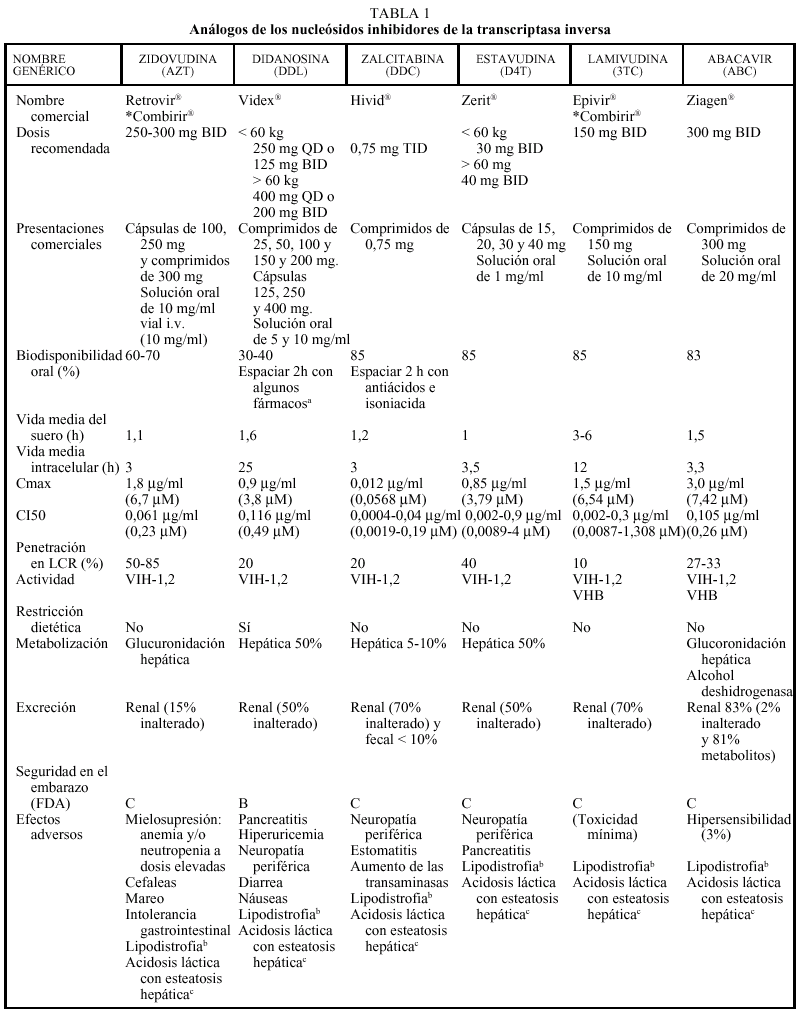
Análogos de nucleósidos (tabla 1)
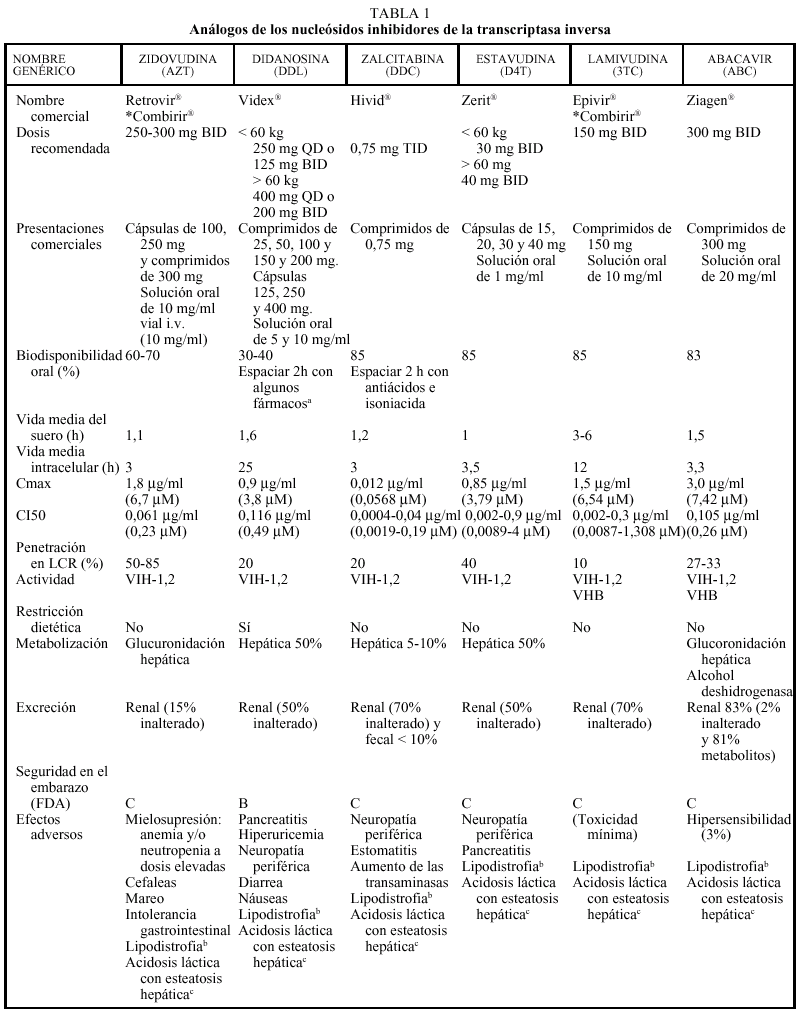
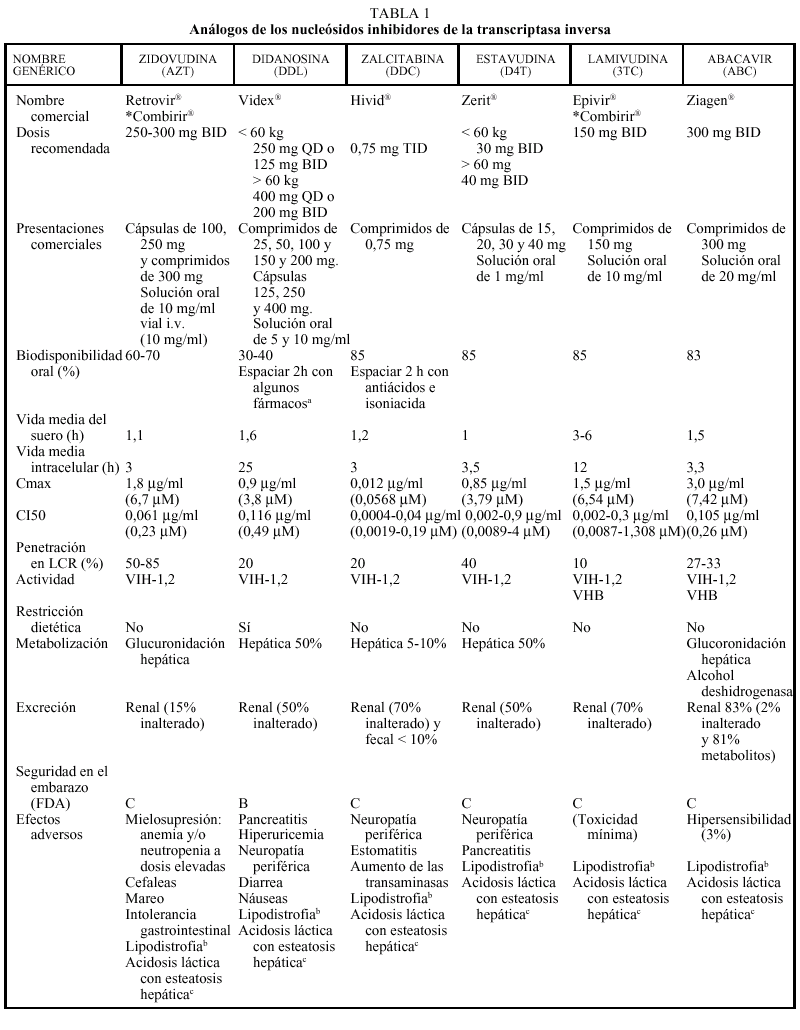
Los análogos de nucleósidos inhibidores de la transcriptasa inversa (TI) fueron el primer grupo de fármacos antirretrovirales utilizados para el tratamiento de la infección por el VIH con la introducción de la zidovudina (AZT) en 1987. En la actualidad hay cinco comercializados en nuestro país que, por orden cronológico, son: AZT, didanosina (ddI), zalcitabina (ddC), estavudina (d4T), lamivudina (3TC) y abacavir (ABC).
Estructura química
EL AZT, ddC y d4T son 2'-3' dideoxinucleósidos pirimidínicos, el ddI es un 2'-3' dideoxinucleósido purínico, el 3TC es un enantiómero negativo (cis) de la 2'-deoxi-3'-tiacitidina y el abacavir es un análogo nucleósido de estructura carbocíclica. Todos ellos nece sitan para su activación sufrir tres fosforilizaciones mediadas enzimáticamente por cinasas intracelulares.
Mecanismo de acción
El mecanismo de acción de los análogos nucleósidos inhibidores de la TI es doble: por un lado, inhiben la TI viral de forma competitiva al unirse a la enzima de una forma más natural que los sustratos nucleósidos naturales y, por otro lado, actúan finalizando la síntesis de la cadena de ADN proviral.
Actividad in vitro
Todos los análogos nucleósidos son activos frente al VIH-1 y el VIH-2; sin embargo, pueden destacarse algunas particularidades: el AZT y el d4T son más activos frente a células en fase de replicación, mientras que el 3TC y el ddI presentan una actividad más intensa en células mononucleares en reposo. El 3TC es activo frente al VHB, ante el que también ha demostrado actividad el ABC. De las distintas combinaciones entre análogos nucleósidos está contraindicada la de AZT + d4T por antagonismo, el AZT compite con el d4T por las cinasas celulares inhibiendo la fosforilización del d4T (la timidina cinasa tiene 600 veces más afinidad por AZT que por d4T).
Farmacocinética
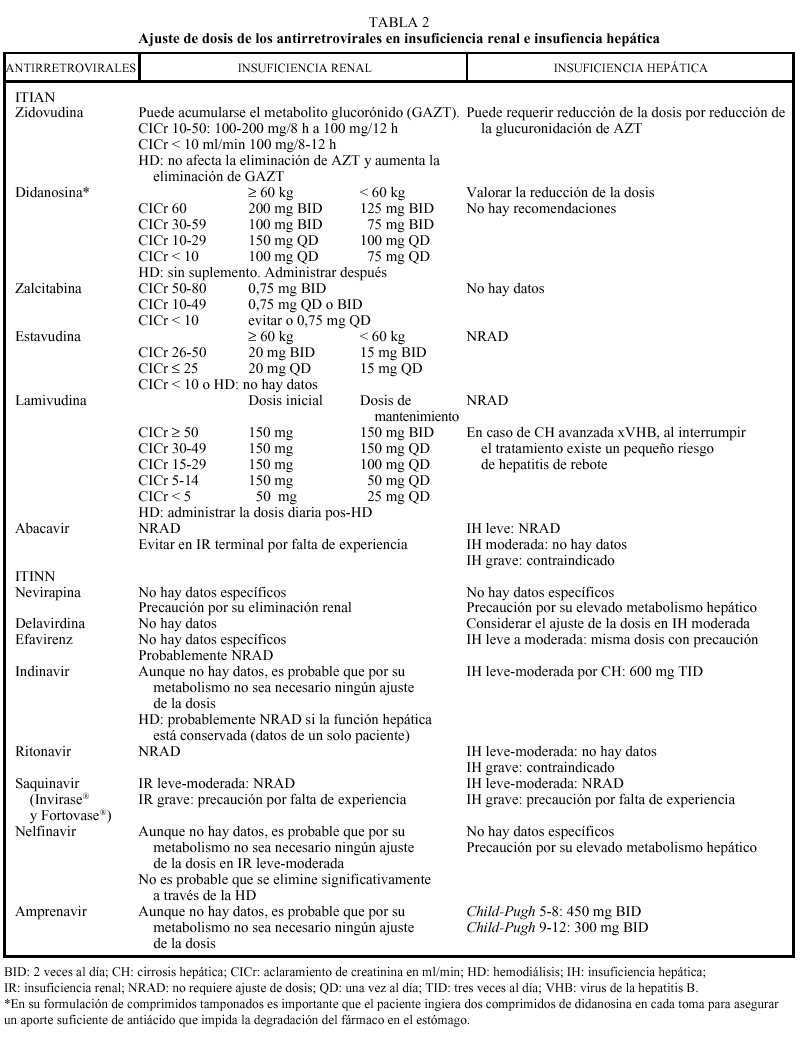
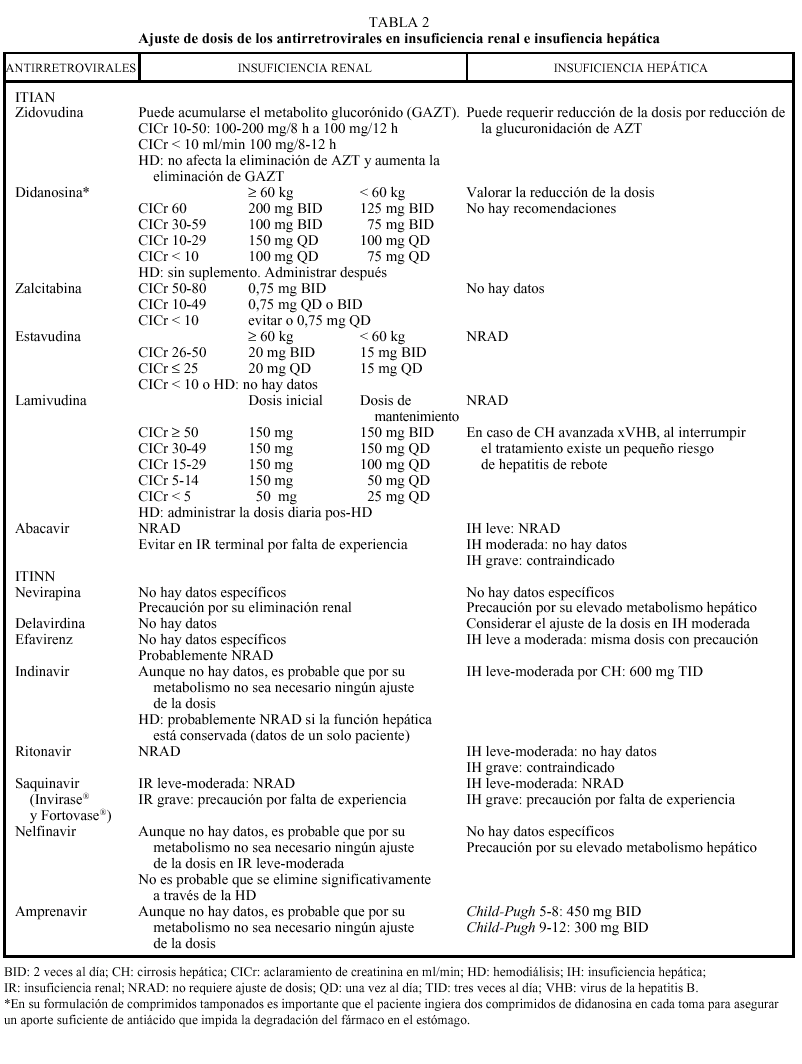
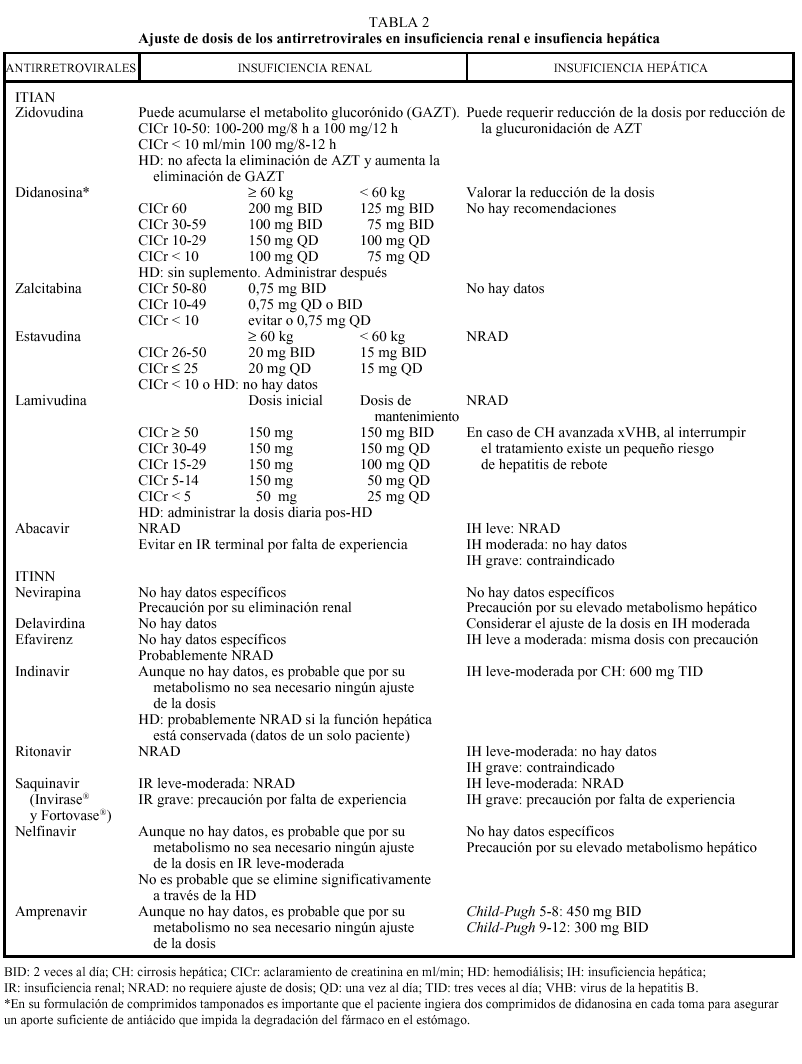
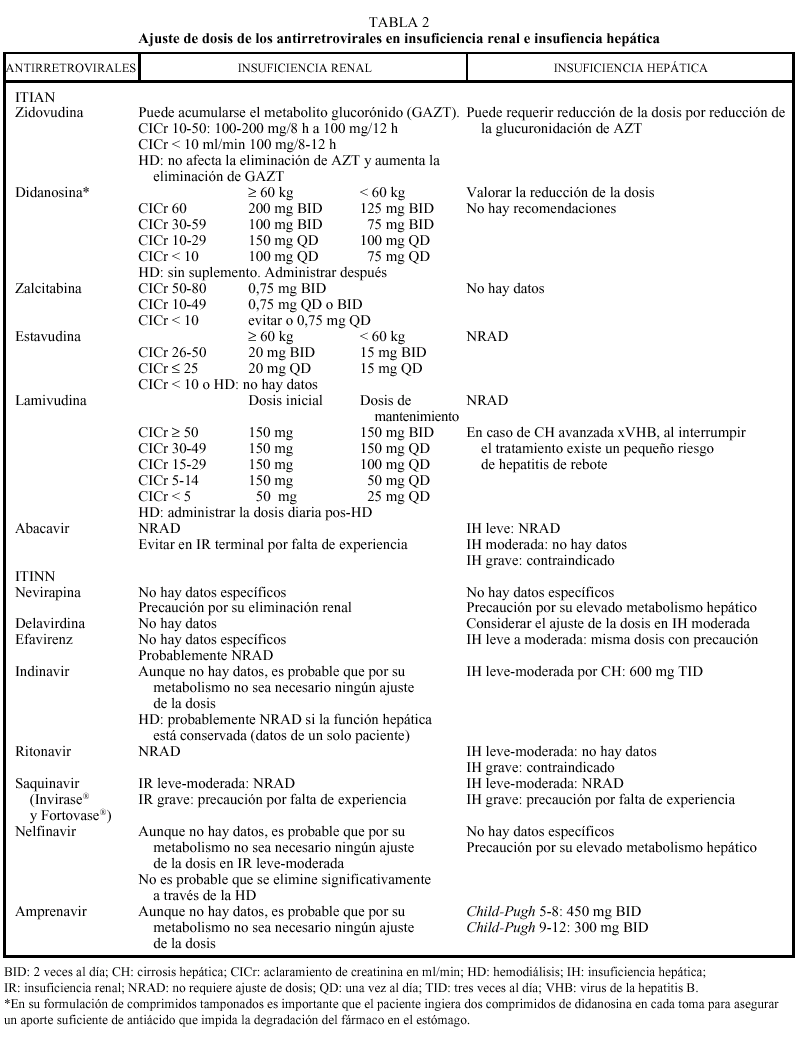
Los análogos de los nucleósidos se pueden administrar con alimentos salvo el ddI que debe administrarse en ayunas. Los análogos de nucleósidos presentan una vida media plasmática corta; sin embargo, su vida media intracelular es más prolongada, especialmente en el caso de ddI y 3TC. DdI y d4T se metabolizan, en parte, en el hígado, al igual que AZT y ABC, que sufren glucuronidación hepática, mientras que ddC y 3TC se eliminan en un elevado porcentaje inalterados por vía renal. La posología en pacientes con insuficiencia renal o hepática se describe en la tabla 2.
Resistencias
La rápida aparición de resistencia que se genera frente al 3TC unas 12 semanas contrasta con el tiempo más prolongado que requieren el resto de análogos para que se generen cepas resistentes a los mismos entre 6 y 12 meses. Aunque existen con frecuencia patrones de resistencia cruzada entre los distintos análogos nucleósidos, las mutaciones principales que se asocian a resistencias son diferentes: AZT (215,70,41), ddI (74), ddC (69), 3TC (184), d4T (75), ABC (65,74,184). Hay que destacar los siguientes aspectos: la mutación 184 revierte la resistencia frente al AZT por las mutaciones 215 y 41; la mutación 75 confiere resistencia frente al d4T in vitro, aunque su significado clínico es desconocido; múltiples mutaciones frente al AZT se asocian a resistencia frente al d4T; las mutaciones E44D y V118I parecen conferir una disminución moderada (en torno a 10 veces) de la sensibilidad a la lamivudina; la mutación 184 parece estar asociada a una disminución del fitness viral, a un aumento en la fidelidad de la TI viral y a una pirofosforilasa defectiva (revierte la resistencia a AZT); están apareciendo más frecuentemente mutaciones que confieren resistencia múltiple a los análogos de nucleósidos, como la 151 y la inserción de una cadena de 6 pares de bases entre los codones 69 y 70.
Efectos secundarios
Los principales efectos secundarios a los análogos nucleósidos según los diferentes fármacos son: AZT (mielotoxicidad-anemia-neutropenia, que ocurren a dosis elevadas, el doble de las actualmente utilizadas, por lo que no se observan con tanta frecuencia como antes,
cefalea, astenia o intolerancia gastrointestinal), ddI (pancreatitis, intolerancia gastrointestinal, neuropatía periférica, diarrea), ddC (polineuropatía periférica, estomatitis) d4T (neuropatía periférica), 3TC (escasa toxicidad), ABC (hipersensibilidad en el 3% de los pacientes). Mención especial merece el cuadro de hipersensibilidad producido por ABC, que se caracteriza por un exantema cutáneo, fiebre y afección del estado general; aunque raro aparece en sólo un 2-3% de los casos, en caso de padecerlo obliga a suspender de forma definitiva el ABC ya que su reintroducción podría provocar un cuadro grave e incluso la muerte del paciente. Raramente se han descrito casos de esteatosis hepática y acidosis láctica, que clínicamente se manifiesta por malestar general, dolor y distensión abdominal, náuseas y vómitos y, desde el punto de vista analítico, por una alteración de la bioquímica hepática así como una elevación del anión gap y de los valores de lactato. No es recomendable la asociación de ddI y ddC por el alto riesgo de toxicidad (neuropatía periférica). Recientemente, se está implicando también al grupo de los análogos de nucleósidos con un fenómeno de alteración en la distribución de la grasa del organismo conocido como lipodistrofia (este monográfico tiene un capítulo dedicado especialmente a este tema).
Interacciones farmacocinéticas y farmacodinámicas (tabla 1)
El ddI, en su formulación de comprimidos tamponados, presenta interacciones en la absorción con diferentes fármacos, especialmente quinolonas (ciprofloxacino, ofloxacino), otros antirretrovirales (indinavir, delavirdina), tuberculostáticos (etambutol, isoniacida, rifampicina), antifúngicos (itraconazol, ketoconazol) y tetraciclinas. Se recomienda administrar estos fármacos 2 h antes del ddI. La nueva formulación en cápsulas entéricas no es de esperar que presente este tipo de interacciones. Como ya se ha comentado anteriormente, algunas asociaciones de análogos, como d4T + AZT o ddC + 3TC, están contraindicadas por antagonismo en la activación intracelular. Otras asociaciones están contraindicadas por el incremento del riesgo de toxicidad, como ddI + ddC o ddC con disulfiram, metronidazol, pentamidina intravenosa y vincristina por el aumento del riesgo de neuropatía periférica. Al asociar 3TC con cotrimoxazol a altas dosis puede reducirse la eliminación de 3TC, ya que ambos fármacos se eliminan por vía renal y, por ello, debe evitarse esta combinación. No hay problema si el cotrimoxazol se administra a dosis profilácticas. En general, las interacciones de los análogos con metadona son poco relevantes, a excepción del ddI que puede requerir un aumento de la dosis.
Análogos de los nucleótidos
Los análogos de los nucleótidos constituyen un grupo de fármacos antirretrovirales de relativamente reciente aparición, no comercializados por ahora, que se encuentran en fase de investigación clínica. En este grupo se incluyen dos fármacos: el adefovir y el tenofovir DF, profármaco oral del PMPA. El adefovir esta disponible a través de un estudio expandido, mientras el tenofovir está actualmente empleándose en estudios en fase III. Muy recientemente, la FDA (noviembre de 1999) no ha aprobado la comercialización del adefovir aduciendo que aún no existen datos suficientes de eficacia y seguridad a dosis de 60 mg/día.
Estructura química
Adefovir (PMEA, 9-[-2-fosfometoxietil]-adenina) y tenofovir DF (BisPOC PMPA) son análogos de nucleótidos acíclidos monofosfato que al no requerir la fosforación inicial dependiente de la nucleósido-cinasa ce lular para ejercer su acción pueden, a diferencia de los análogos nucleósidos que sí la requieren, ampliar su acción sobre distintos grupos celulares como monocitos, macrófagos y células T, tanto en reposo como activados.
Mecanismo de acción
Actúan al inhibir de forma selectiva la polimerasa viral, compitiendo con la desoxiadenosina trifosfato en su incorporación a la molécula de ADN e interrumpiendo el crecimiento de la misma.
Actividad in vitro
El adefovir ha demostrado ser activo frente a múltiples retrovirus, entre los que destacan el VIH-1 (CI50 0,3-14,5 mg/ml) y el VIH-2, así como frente a distintos virus ADN como el hepadnavirus (VHB) y los virus de la familia herpes (HSV-1, HSV-2, CMV, VEB, herpesvirus tipo 6). El tenofovir tiene actividad frente al VIH-1 y el VIH-2, el SIV y el virus causante del sarcoma murino-Monoley.
Farmacocinética
El adefovir tiene una baja biodisponibilidad. Para aumentarla, se administra como profármaco (adición de dos grupos piválicos) el adefovir dipivoxilo (bis-POM PMEA), que tiene una biodisponibilidad del 40% con la comida. Tras una dosis de 125-250 mg, su Cmax, Tmax y ABC son de 0,21 mg/ml, 2 h y 3,22 mg/h/ml, respectivamente. La vida media plasmática es de 1,5 h y la intracelular de 16-18 h, lo que permite su dosificación una vez al día. No sufre metabolismo hepático y se elimina por vía renal sin cambios en un 90% (secreción tubular activa).
El tenofovir presenta también una limitada biodisponibilidad oral, por lo que se administra como profármaco (derivado estérico) el tenofovir disoproxil fumarato (tenofovir DF), que tiene una biodisponibilidad del 40% con la comida. Tras una dosis única de 300 mg de tenofovir DF se alcanzó una concentración plasmática máxima de 362 ng/ml. La vida media en fase terminal estimada fue igual o superior a 17 h. El tenofovir se elimina inalterado por la orina (filtración glomerular y secreción tubular activa).
Resistencias
En cuanto a los mecanismos de resistencia, en lo referente al adefovir, estudios in vitro han demostrado que las mutaciones K65R y K70E del gen de la TI confieren un descenso de la sensibilidad al adefovir de 16 y 9 veces, respectivamente; la mutación M184V ha demostrado aumentar entre 3 y 4 veces la sensibilidad a adefovir. En cuanto al tenofovir, una mutación en la posición 65 disminuye entre 3 y 4 veces la sensibilidad frente al mismo; la mutación en el codón 215 (resistencia a AZT) disminuye algo menos de 2 veces la sensibilidad a tenofovir, mientras que la 184 (asociada con resistencia a 3TC) aumenta la sensibilidad a tenofovir unas 2 veces.
Efectos secundarios
Los efectos secundarios más frecuentes descritos con los análogos nucleótidos son de tipo gastrointestinal (vómitos, sensación de malestar abdominal y diarrea); respecto al adefovir merece una mención especial la afección renal, que se asocia al mismo en forma de acidosis tubular renal (ATR) tipo 2 y que se ha constatado en el 20% de los casos a los 6-12 meses, o la insuficiencia renal reversible. En algunos casos se ha descrito una leve citólisis hepática, exantema, neutropenia, neuropatía e hiperamilasemia. No se han descrito efectos adversos graves con el tenofovir, siendo los efectos adversos menores descritos la cefalea, el mareo y la astenia. La afección renal es excepcional.
Posología
La posología del adefovir será, probablemente, de 60 mg/día y la del tenofovir de 300 mg/día, ambos en dosis única diaria sin restricción dietética.
Interacciones farmacocinéticas y farmacodinámicas
El adefovir no tiene interacciones significativas con otros antirretrovirales. Sin embargo, el profármaco del adefovir (adefovir dipivoxilo) genera en su metabolismo ácido piválico, que se esterifica con la carnitina libre para su excreción renal. La carnitina es necesaria para el transporte de ácidos grasos a través de la membrana mitocondrial para su posterior oxidación. Su déficit se asocia a miopatía, cardiopatía y encefalopatía. A pesar de que este tratamiento no genera déficit de carnitina, para mayor seguridad se administran 500 mg/día de carnitina. La asociación de adefovir con otros fármacos nefrotóxicos aumenta el riesgo de toxicidad renal.
No análogos de los nucleósidos (tabla 3)
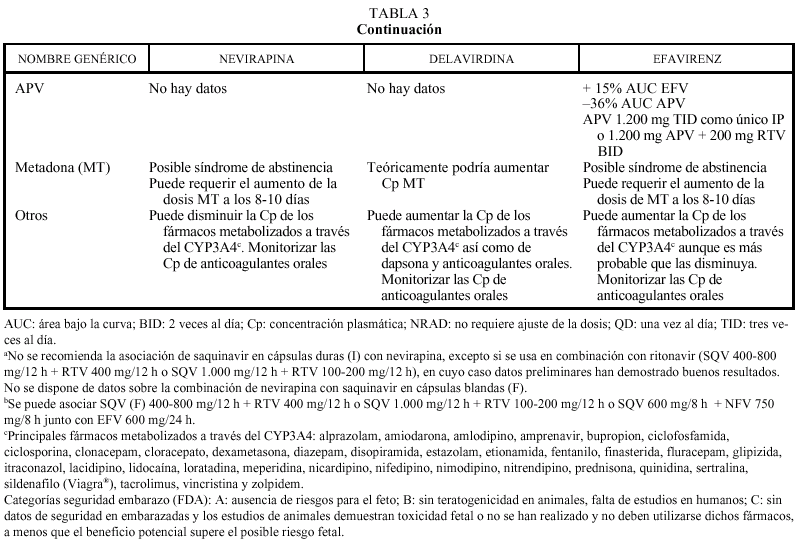
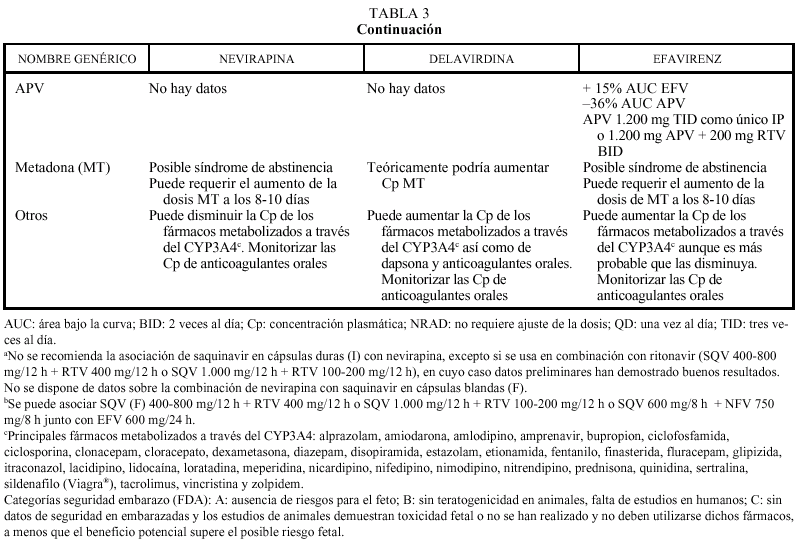
El grupo de los inhibidores de la TI no análogos de los nucleósidos, actualmente aprobados por la FDA y la EMEA y disponibles en nuestro país, está formado por tres fármacos: delavirdina (DLV), nevirapina (NVP) y efavirenz (EFV). La delavirdina está disponible en la actualidad a través de un Programa de Acceso Expandido (NPSP-3331).
Estructura química
Los no análogos de nucleósidos son compuestos de estructura química compleja y heterogénea. Desde el punto de vista molecular, NVP es un derivado de la dipiridodiazepinona, EFV es (S)-6-cloro-4-(ciclopentanil)-1,4-dihidro-4-(trifluorometil)-2H-3,1-benzoxacin-2-ona, y DLV pertenece a la familia de las bis (hetaryl) piperazinas.
Mecanismo de acción
A diferencia de los análogos de los nucleósidos, los no análogos de nucleósidos no requieren ser fosforilizados para ejercer su acción. Son activos frente al VIH-1 pero no frente al VIH-2 ni frente a hepadnavirus ni virus del grupo herpes. Actúan a través de un bloqueo de la actividad de la ADN polimerasa viral dependiente del ARN y del ADN, uniéndose de forma directa a la TI por un sitio distinto del que se une al sustrato dNTP, a diferencia de los análogos de los nucleósidos que se unen a ésta de forma competitiva. A pesar de actuar al mismo nivel que los análogos de nucleósidos, ambos actúan inhibiendo la TI viral, su combinación es generalmente sinérgica o al menos aditiva.
Farmacocinética
Los no análogos presentan una buena biodisponibilidad oral y, a diferencia de los análogos, tienen una semivida plasmática muy larga (a excepción de delavirdina), lo que permite su administración una vez al día; DLV requiere medio ácido para su absorción, por lo que ésta puede verse afectada por la presencia de antiácidos (v. apartado interacciones); NVP y EFV presentan una buena penetración en LCR.
La metabolización de todos los no análogos de los nucleósidos es hepática y a través del CYP3A4, sistema enzimático del citocromo P450. La posología en pacientes con insuficiencia renal o hepática se describe en la tabla 2.
Resistencias
La resistencia cruzada entre los distintos no análogos de los nucleósidos es muy frecuente; además, estos fármacos tienen una barrera genética muy baja, por lo que las resistencias aparecen con extremada rapidez. Mutaciones únicas confieren un alto nivel de resistencia, de forma que una mutación en la posición 181 confiere un aumento de la resistencia a NVP y DLV de unas 1.000 veces. Las mutaciones que confieren resistencia a los no análogos de los nucleósidos se encuentran entre las posiciones 100-108 y 179-190. La mutación más frecuente que aparece cuando se desarrolla resistencia a EFV se localiza en la posición 103, y por sí sola confiere un aumento de la resistencia al mismo de unas 20 veces; esta mutación confiere igualmente un alto nivel de resistencias a NVP y DLV. Otras mutaciones que se asocian a un alto nivel de resistencias a los no análogos de los nucleósidos son la 106, 108 y 190 para la NVP y la 188 y 190 para EFV.
Efectos secundarios
El exantema es un efecto adverso común a todos los no análogos de los nucleósidos, siendo más frecuente, por este orden, en DLV, NVP y EFV; su aparición no siempre obliga a la suspensión definitiva del fármaco. Otro efecto común es la elevación de las transaminasas, que ocasionalmente puede obligar a suspender el fármaco (si la elevación de las transaminasas suepera más de 5 veces el límite alto de la normalidad). El EFV se asocia en un 20-40% a alteraciones neuropsiquiátricas en forma de mareos, inestabilidad, sueños intensamente vívidos y, más raramente, cuadros depresivos e ideaciones suicidas. Con EFV se ha detectado teratogenicidad en monos, por lo que es un fármaco no recomendado en el embarazo.
Interacciones farmacocinéticas y farmacodinámicas
El DLV requiere un medio ácido para su absorción, debiendo separarse su toma de la del ddI (o cualquier otro fármaco que contenga antiácidos) por lo menos 1 h. Su asociación a fármacos como ranitidina y omeprazol está contraindicada. Los no análogos de los nucleósidos actúan de forma diversa sobre la isoenzima CYP3A4: DLV la inhibe, NVP la induce y EFV puede actuar de forma indistinta, aunque predomina el efecto inductor. Por tanto, puede requerirse un ajuste de la dosis si se administran otros fármacos que son sustratos del citocromo P450 o que lo inhiben o inducen. Delavirdina, por su efecto inhibidor del metabolismo, puede aumentar las concentraciones plasmáticas de algunos fármacos aumentando su toxicidad y, por ello, está contraindicada su asociación con algunos antihistamínicos H1 (terfenadina, astemizol), benzodiacepinas (midazolam, triazolam), derivados de la ergotamina, pimozida, cisaprida y estatinas (sí puede administrarse pravastatina, porque se elimina fundamentalmente por vía renal). Por otro lado, los fármacos que inducen el metabolismo hepático (carbamacepina, fenitoína, fenobarbital, rifabutina o rifampicina) pueden reducir las concentraciones plasmáticas de delavirdina hasta valores subterapéuticos y no deben administrarse conjuntamente. Efavirenz puede actuar reduciendo el metabolismo de otros fármacos (por lo que las contraindicaciones serían similares a delavirdina) o aumentándolo y reduciendo su eficacia, al igual que nevirapina (tabla 3). Un ejemplo sería la reducción de la eficacia de los anticonceptivos orales cuando se administra nevirapina. En otros casos será necesario ajustar las dosis, como ocurre con frecuencia al combinar no nucleósidos con antimicobacterianos e inhibidores de la proteasa del VIH. Con rifampicina puede administrarse efavirenz (se debe valorar el aumento de dosis a 800 mg al día). Rifabutina puede asociarse a nevirapina, sin necesidad de ajustar las dosis, o con efavirenz (aumentando rifabutina a 450 mg/día). En cuanto a los inhibidores de la proteasa, debe valorarse el aumento de la dosis de indinavir a 1.000 mg/8 h con nevirapina y efavirenz, mientras que con delavirdina se reducirá la dosis de indinavir a 600 mg/8 h. Ritonavir y nelfinavir se pueden asociar a todos los no nucleósidos, aunque en el caso de delavirdina no hay datos sobre ajuste de la dosis (tabla 2). Saquinavir solamente puede asociarse a nevirapina o efavirenz combinándolo con ritonavir (de otro modo no alcanzaría valores plasmáticos suficientes). Amprenavir puede asociarse a efavirenz ajustando la dosis (tabla 3). Otra interacción con relevancia clínica es la que se produce con metadona. Nevirapina y efavirenz pueden reducir sus concentraciones plasmáticas e inducir un síndrome de abstinencia (habitualmente a los 8-10 días), por lo que se deberá aumentar la dosis de metadona hasta obtener el efecto deseado. Por último, en pacientes que reciban anticoagulantes, deberá monitorizarse estrictamente el tiempo de protrombina, sobre todo al iniciar o finalizar un tratamiento antirretroviral con no análogos.
Inhibidores de la proteasa (tabla 4)
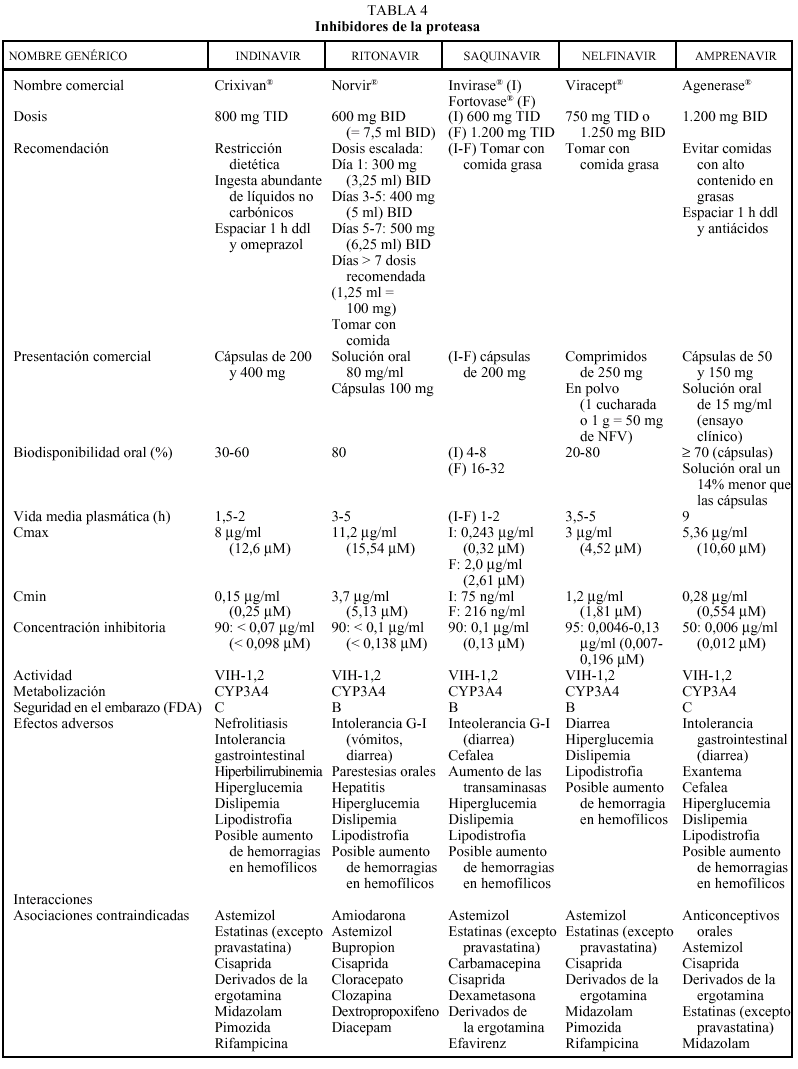
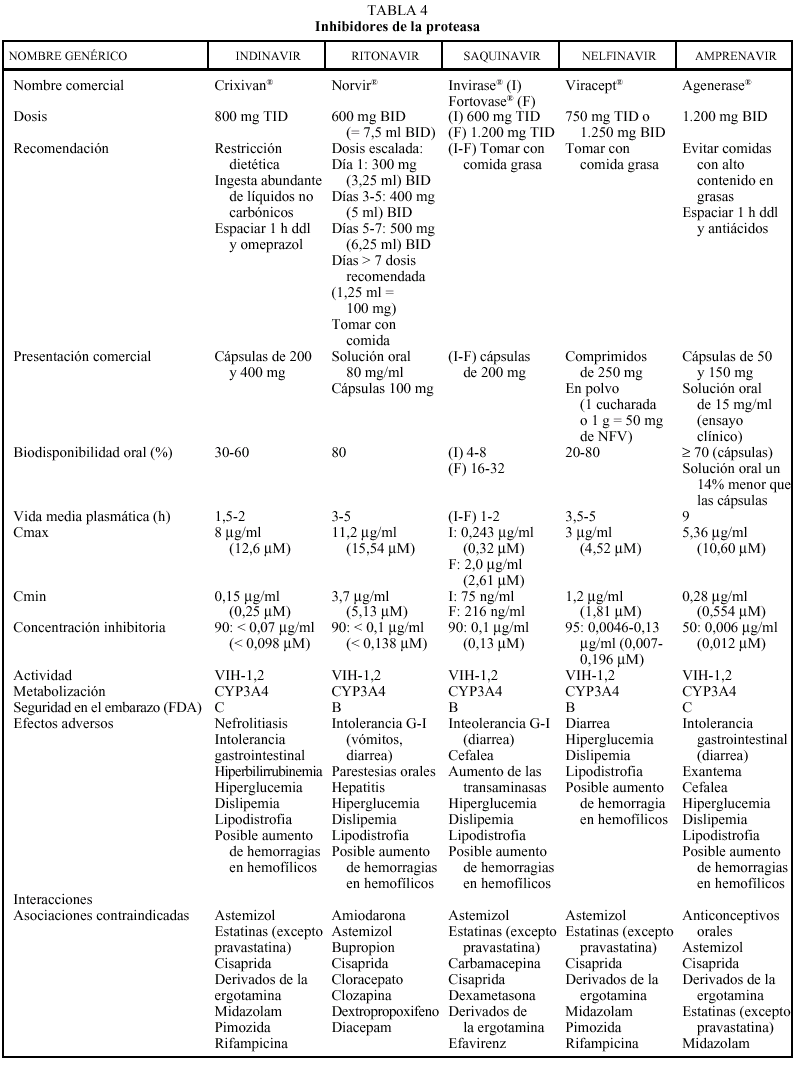
Los inhibidores de la proteasa (IP) revolucionaron, con su aparición en 1996, el tratamiento de la infección por el VIH, provocando una disminución espectacular de la morbimortalidad. En la actualidad existen en nuestro país comercializados cuatro IP: saquinavir (SQV) (en dos formulaciones, cápsulas de gelatina dura y, cápsulas de gelatina blanda), indinavir (IDV), ritonavir (RTV) y nelfinavir (NFV); se dispone del amprenavir (APV) a través de un ensayo clínico en fase III (PROA/3006).
Estructura química
Los IP disponibles en la actualidad son todos moléculas complejas de tipo peptidomimético que tienen selectividad frente a la proteinasa del VIH-1 y el VIH-2.
Mecanismo de acción
Los IP actúan inhibiendo la proteasa viral, enzima que cataliza la escisión de las poliproteínas gag y gag-pol en proteínas estructurales clave y enzimas de replicación del VIH necesarias para la maduración y la proliferación viral.
Farmacocinética
Los IP presentan una farmacocinética poco favorable, en general, con una baja biodisponibilidad (especialmente saquinavir) y una corta vida media, aunque con una gran potencia intrínseca. Para una óptima absorción, ritonavir y especialmente saquinavir y nelfinavir deben administrarse junto con alimentos, mientras que indinavir se absorbe mejor con el estómago vacío (1 h antes o 2 h después de las comidas). En caso de intolerancia digestiva, indinavir puede tomarse con leche desnatada, zumos, café, té o comida ligera en grasas (tostadas con mermelada, cereales, azúcar); y cuando se utiliza asociado a ritonavir, indinavir puede ser administrado junto con una comida habitual. Todos los IP se metabolizan a través del hígado y principalmente a través de la isoenzima CYP3A4 del citocromo P450, vía que comparten con otros fármacos con los que pueden interaccionar de forma notable, lo que puede obligar a modificaciones importantes en las posologías (v. apartado interacciones). El ajuste de las dosis en pacientes con insuficiencia renal o hepática se describe en la tabla 2.
Resistencias
Los IP presentan una barrera genética elevada para el desarrollo de resistencias, es necesario la acumulación de múltiples mutaciones para que se genere un alto nivel de resistencia fenotípica. Las mutaciones que se desarrollan frente a cada uno de los IP pueden ser primarias o secundarias; las primarias se caracterizan por aparecer en primer lugar y por disminuir la capacidad de unión del fármaco a su enzima diana; las secundarias o accesorias no modifican la sensibilidad pero sí el fitness viral. El grado de resistencia al fármaco suele ser directamente proporcional al número de mutaciones, las mutaciones primarias relacionadas con los distintos IP son: IDV (46,82), SQV (48,90), RTV (82), NFV (30,90) y APV (50,84). La presencia de mutaciones primarias confiere resistencia cruzada entre los IP que las comparten, aunque la existencia de dos o más mutaciones clave o primarias confiere un alto nivel de resistencia cruzada a todos los IP. Amprenavir es el único que no comparte mutaciones, excepto con ritonavir en caso de virus multirresistentes.
Efectos secundarios
Los IP son, en general, un grupo de fármacos relativamente mal tolerados por vía oral. Todos ellos se asocian a efectos secundarios de aparición a corto, medio y largo plazo. Entre los efectos secundarios a corto plazo destacan la nefrolitiasis y la intolerancia gastrointestinal con el IDV, la diarrea con el NFV y la intolerancia gastrointestinal con RTV. Los trastornos metabólicos, así como un fenómeno de redistribución anómala de la grasa del organismo conocido como lipodistrofia, son efectos secundarios que aparecen, a medio y largo plazo, asociados con frecuencia a los IP. De entre los trastornos metabólicos, la hipercolesterolemia, la hipertrigliceridemia y, en menor medida, la hiperglucemia son los más frecuentes. La lipodistrofia asociada a los IP se caracteriza por una lipoatrofia periférica (con pérdida de grasa subcutánea en las piernas, los brazos y los glúteos y facial bolsa de Bichat), que se acompaña de una obesidad central (con acumulación de grasa en el abdomen, los senos y el cuello), a diferencia de la lipodistrofia asociada a los análogos de los nucleósidos que se presenta de forma más característica como una lipoatrofia generalizada.
Interacciones farmacocinéticas y farmacodinámicas
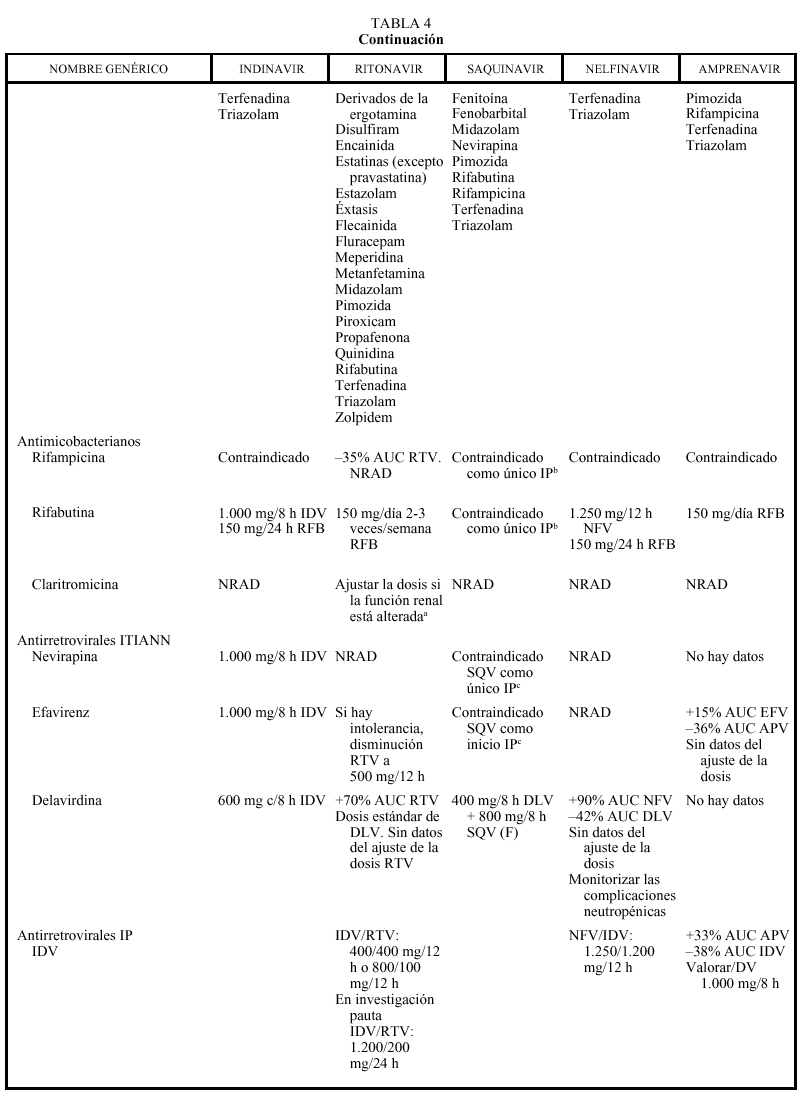
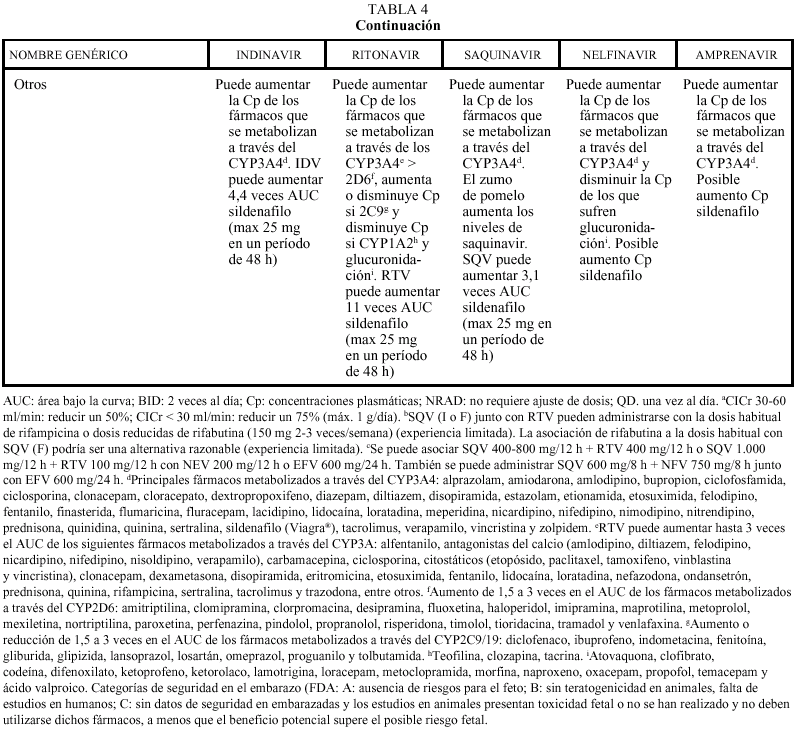
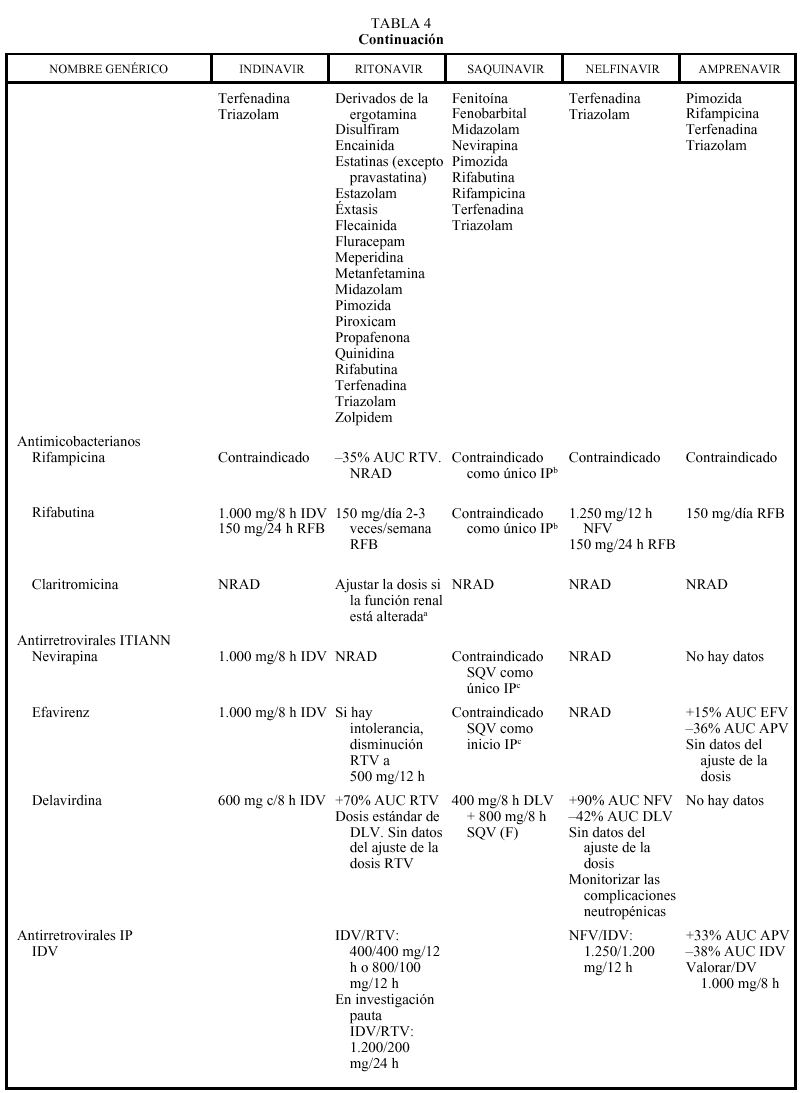
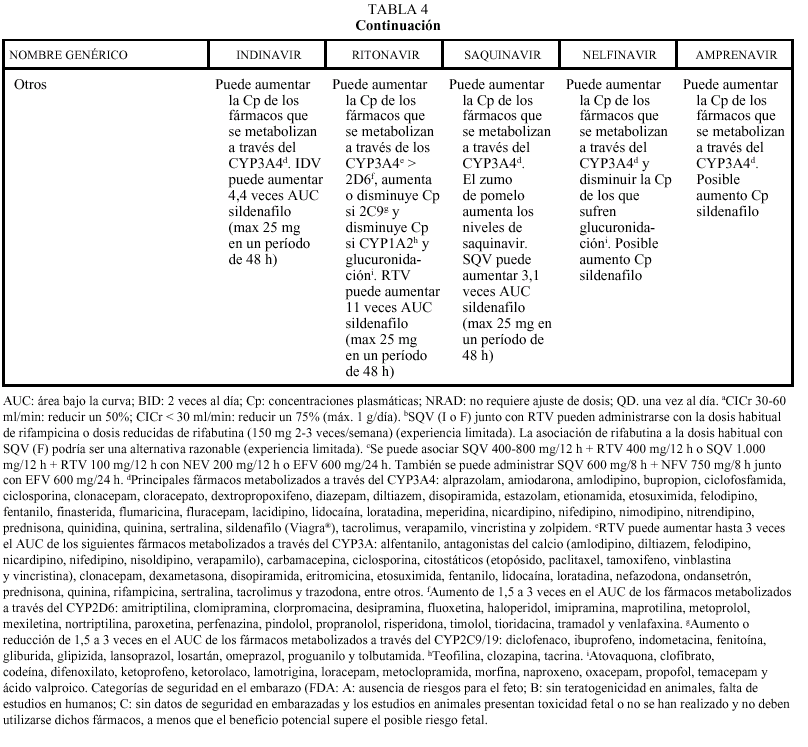
En general, los IP actúan como inhibidores del metabolismo de otros fármacos, aumentando sus concentraciones plasmáticas y su toxicidad: por ello, al igual que ocurría con delavirdina o efavirenz, está contraindicada su asociación con algunos antihistamínicos H1 (terfenadina, astemizol), benzodiacepinas (midazolam, triazolam), derivados de la ergotamina, pimozida, cisaprida y estatinas (a excepción de pravastatina). Ritonavir y nelfinavir en algunas ocasiones también pueden aumentar el metabolismo (p. ej., la glucuronidación). La isoenzima del citocromo P450 que más inhiben los IP es la CYP3A4, y ritonavir es el que presenta un efecto inhibidor más potente (seguido de indinavir, amprenavir, nelfinavir, delavirdina y saquinavir), por lo que es el que tiene un mayor riesgo de interacción (tabla 4). Sin embargo, no siempre las interacciones tienen un efecto perjudicial; en este sentido, la importante acción inhibidora de RTV sobre la isoenzima CYP3A4 va a permitir, al emplearlo conjuntamente con otros IP, la administración de dosis más reducidas y espaciadas de éstos (IDV 800 mg + RTV 100 mg BID; IDV 400 mg + RTV 400 mg BID; SQV-CGD 400 mg + RTV 400 mg BID). El empleo de RTV a dosis bajas (100-200 mg en la llamada «babydose») permite disminuir el número de comprimidos y ampliar el intervalo posológico de otros IP, como IDV o SQV (tabla 4). Los IP presentan interacciones importantes entre sí que obligan a ajustar la dosis cuando se combinan entre ellos (tabla 4). En pacientes con tuberculosis en tratamiento con IP, rifampicina puede adminitrarse solamente si reciben ritonavir (solo o asociado a saquinavir). Rifabutina puede combinarse con cualquiera de los IP, a excepción de saquinavir en cápsulas de gelatina dura (que sólo se asociará a rifabutina combinado con ritonavir). Debido al efec-
to inhibidor de los IP, la dosis de rifabutina deberá reducirse a 150 mg al día, y en las combinaciones que contengan ritonavir, solamente se administrará rifabutina, 150 mg 2-3 veces por semana. Debido al efecto inductor de rifabutina deberán aumentarse las dosis de in-
dinavir y nelfinavir a 1.000 mg/8 h. Respecto a las interacciones con los no nucleósidos, como ya se ha comentado anteriormente, debe valorarse el aumento de la dosis de indinavir a 1.000 mg/8 h con nevirapina y efavirenz, mientras que con delavirdina se reducirá la dosis de indinavir a 600 mg/8 h. Ritonavir y nelfinavir se pueden asociar a todos los no nucleósidos, aunque en el caso de delavirdina no hay datos sobre ajuste de la dosis (tabla 4). Saquinavir solamente puede asociarse a nevirapina o efavirenz combinándolo con ritonavir (de otro modo no alcanzaría valores plasmáticos suficientes). Amprenavir puede asociarse a efavirenz ajustando la dosis (tabla 4). Los pacientes en pauta de deshabituación con metadona pueden requerir un aumento de dosis de metadona con ritonavir. Con nelfinavir, a pesar de que en algunas personas pueden reducirse las concentraciones de metadona a la mitad, no parece que se requieran grandes ajustes en la dosis, al igual que ocurre con indinavir, saquinavir y amprenavir. No hay datos sobre amprenavir. Se recomienda mucha precaución al administrar IP a pacientes en tratamiento anticoagulante ya que, aunque por su efecto inhibidor del metabolismo sería de esperar un aumento del efecto anticoagulante, existen casos descritos de reducción del efecto anticoagulante con requerimiento de aumento de la dosis de acenocumarol y warfarina al asociarlos a RTV, IDV o NFV. Se debe monitorizar estrictamente el tiempo de protrombina, sobre todo al iniciar o suspender
un tratamiento con IP. Se recomienda también mucha precaución con otros fármacos de estrecho margen terapéutico y, en general, con todos los que sufran un elevado metabolismo hepático, como por ejemplo sildenafilo (Viagra®).
Bibliografía recomendada
Carpenter CCJ, Cooper DA, Fischl MA, Gatell JM, Gazzard BG, Hammer SM et al. Antiretroviral Therapy in Adults. Updated Recommendations of the International AIDS Society-USA Panel. JAMA 2000; 283: 381-390.
CDC. Updated guidelines for the use of rifabutin or rifampin for the treatment and prevention of tuberculosis among HIV-infected patients taking protease inhibitors or nonnucleoside reverse transcriptase inhibitors. MMWR 2000; 49: 185-189.
Gatell JM, Clotet B, Podzamczer D, Miró JM, Mallolas J. Guía práctica del sida. Clínica, diagnóstico y tratamiento (6.a ed.). Barcelona: Masson, 2000.
Guidelines for the Use of Antiretroviral Agents in HIV-infected Adults and Adolescents. Departamento de Salud y Servicios Humanos de los Estados Unidos (DHHS) y la fundación Henry J. Kaiser Family. Edición de 28 enero de 2000 (http//: www.hivatis.org).
Hirsch MS, Brun-Vezinet F, D'Aquila RT, Hammer SM, Johnson VA, Kuritzkes DR et al. Antiretroviral Drug Resistance Testing in Adult HIV-1 Infection: Recommendations of an International AIDS Society-USA Panel. JAMA 2000; 283: 2417-2426.
Jayasekara B, Aweeka FT, Rodríguez R, Kalayjian RC, Humphreys MH, Gambertoglio JG. Antiviral therapy for HIV patients with renal insuficiency. J Acquir Immune Defic Syndr Hum Retrovirol 1999; 21: 384-395.
Mensa J, Gatell JM, Jiménez de Anta MT, Prats G. Guía de terapéutica antimicrobiana. (10.a ed.). Barcelona: Masson, 2000.
Miró JM, Antela A, Arrizabalaga J et al. Recomendaciones de GESIDA/Plan Nacional sobre el Sida respecto al tratamiento antirretroviral en pacientes adultos infectados por el VIH en el año 2000 (primera parte). Enferm Infecc Microbiol Clin 2000; 18: 329-351.
Miró JM, Antela A, Arrizabalaga J et al. Recomendaciones de GESIDA/Plan Nacional sobre el Sida respecto al tratamiento antirretroviral en pacientes adultos infectados por el VIH en el año 2000 (segunda parte). Enferm Infecc Microbiol Clin 2000; 18: 396-412.
Tuset M, Miró JM, Codina C. Interacciones medicamentosas de interés en la terapia del VIH. En: González-García J, Moreno S y Rubio R, editores. Infección por VIH-1998. Madrid: Doyma, 1998: 281-354.
Tuset M, Miró JM, Codina C, Martínez M, Del Cacho E, Ribas J. Interacciones entre los antirretrovirales y los fármacos utilizados en el tratamiento de la tuberculosis. Enf Emerg 2000; 2: 28-44.