














y el hospedador en un doble contexto. Por una parte, se ha de considerar la interacción entre un virus y una célula; en este «microcosmos» es importante analizar y comprender los mecanismos de adaptación del virus a su célula diana. Existe, sin embargo, un segundo nivel de complejidad, en el que se enfrentan poblaciones virales formadas por miles de millones de partículas y un sistema inmune dotado de numerosos mecanismos de defensa y de amplios repertorios de respuesta antimicrobiana. En este «macrocosmos», el VIH debe ser capaz de adaptarse mediante mecanismos de escape que le permiten eludir la respuesta inmune. En la primera parte de este capítulo se analiza la interacción virus-célula y, en especial, el papel que desempeñan en la inmunopatología de la infección por el VIH los receptores virales y los mecanismos de reactivación del VIH a partir de su estado de latencia. En la segunda parte del capítulo se analizan las interacciones entre la población viral y el sistema inmune. En este apartado se describe el ciclo biológico del VIH in vivo, los mecanismos de destrucción linfocitaria, la respuesta inmune generada por el hospedador y los mecanismos de evasión por parte del virus. Por último, se esbozan las principales preguntas planteadas actualmente en el campo de la inmunopatología del sida.
Ciclo biológico del VIH. Interacción virus-célula
El ciclo biológico del VIH se divide en dos etapas bien diferenciadas (fig. 1): la fase temprana, que culmina con la integración del ADN proviral en el genoma de la célula, y la fase tardía, que supone la transcripción del genoma viral y la generación de una progenie infec-
ciosa1.

Fig. 1.Ciclo biológico del VIH.
Entrada del VIH en la célula. Receptores y tropismo viral
La entrada del VIH en la célula se produce mediante la interacción con dos tipos de receptores. Por una parte, existe un receptor específico y común a todos los subtipos del VIH, la molécula de CD4. Esta proteína está presente en la superficie de los linfocitos T colaboradores y en células de estirpe mononuclear-fagocítica, lo que determina el tropismo viral por estos tipos celulares. Sin embargo, la presencia de la molécula de CD4 es condición necesaria pero no suficiente para permitir la entrada del VIH en la célula (fig. 2) y se requiere, además, la función de otras moléculas. El descubrimiento de que determinados receptores de quimiocinas actúan como correceptores virales tiene una gran trascendencia para comprender distintos aspectos patogénicos de la infección por el VIH2.

Fig. 2.Requerimiento de distintas moléculas para la entrada del VIH en la célula.
Quimiocinas y sus receptores
Las quimiocinas constituyen una familia de mediadores inmunológicos de la que se han descrito al menos 30 proteínas diferentes divididas en tres grupos: C, CC y CXC quimiocinas3. Son moléculas solubles de bajo peso molecular cuya principal función es la de ser mediadores inflamatorios implicados en los procesos de migración y activación leucocitarias. Son producidas por monocitos, polimorfonucleares y linfocitos CD4 y CD8, así como por otras estirpes celulares (fibroblastos, células endoteliales y estromales). Para realizar su función, las quimiocinas interaccionan con distintos receptores situados en la membrana celular que se clasifican de acuerdo con su capacidad para unir las distintas familias de quimiocinas en receptores de tipo C, CC y CXC. Estos receptores tienen una estructura del tipo «siete dominios transmembrana» (7M) y transducen señales de activación mediante su acoplamiento a proteínas G4.
Correceptores del VIH y tropismo viral
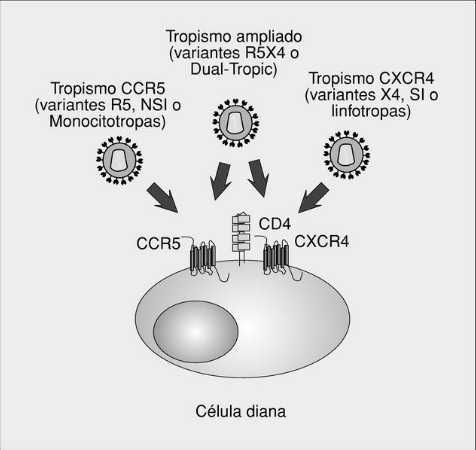
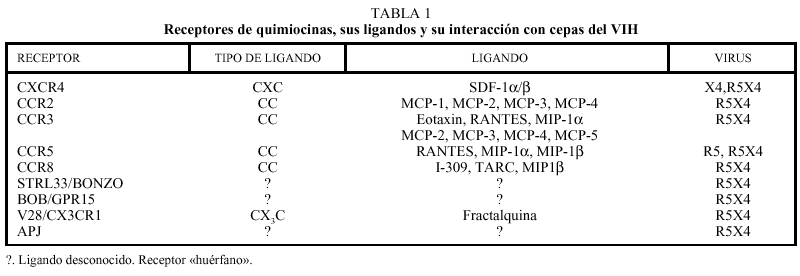
A pesar de que in vitro se ha descrito que muchos receptores de quimiocinas o receptores «huérfanos» pertenecientes a la familia de receptores 7M son capaces de actuar como correceptores del VIH (tabla 1), in vivo probablemente existen sólo dos receptores mayores, que son las moléculas CCR5 y CXCR4. El primero une las CC-quimiocinas RANTES, MIP-1* y MIP-1ß y es el principal receptor de las cepas monocitotropas (previamente denominadas NSI o no sincitiales, actualmente denominadas R5) (fig. 3). El receptor CXCR4 tiene como ligando natural la quimiocina SDF1 y es el principal receptor de las cepas denominadas linfotropas (SI o sincitiales, en la actualidad, denominadas X4). La conformación de distintas regiones de la gp120, especialmente el bucle V3, condiciona el tropismo de los distintos aislados virales. Además de los virus con un tropismo estricto por CCR5 o CXCR4, se han descrito variantes virales capaces de unirse a otros correceptores, como CCR2 o CCR3, y aislados capaces de entrar en la célula a través de múltiples correceptores (cepas de tropismo dual o ampliado, denominadas R5X4)5.
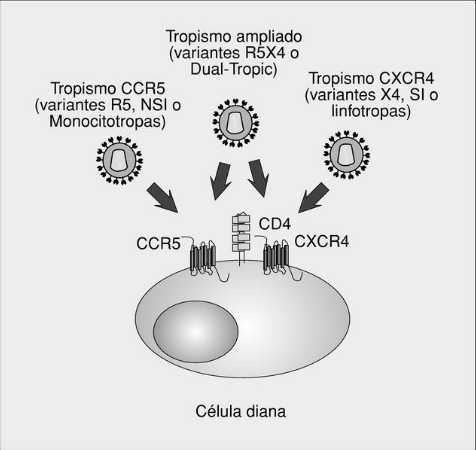
Fig. 3.Correceptores del VIH y tropismo viral.

Correceptores y resistencia a la infección por el VIH
Las quimiocinas que se unen a CCR5 y CXCR4, muy especialmente RANTES y SDF, son capaces de inhibir la infección por el VIH (tabla 1) y con toda probabilidad estos fenómenos constituyen un potente mecanismo antiviral in vivo. Por otra parte, la caracterización de los correceptores del VIH y sus ligandos naturales ha permitido definir una serie de variantes genéticas que se asocian con resistencia a la infección o con una progresión más o menos rápida de la enfermedad (tabla 2)6.

Mecanismo de entrada del VIH en la célula
Una vez concluido el proceso de unión de la gp160 a la molécula de CD4, se producen una serie de cambios conformacionales inducidos por la interacción sucesiva con sus receptores (fig. 4). Desde el punto de vista estructural, la gp160 es una proteína flexible que presenta distintas conformaciones: en su forma nativa o precursora los dominios expuestos se unen a los dominios D1 y D2 de CD4 con alta afinidad7. Esta interacción permite un desplegamiento de la gp160 que expone el dominio V3 y regiones adyacentes que interaccionarán con los correceptores de quimiocinas. Esta segunda interacción entre el asa V3 de la gp120 y el correceptor probablemente induce nuevos cambios en la estructura espacial de la gp120 que expone el dominio N-terminal de la gp41. Esta región contiene el denominado «péptido de fusión», una región altamente hidrofóbica que una vez anclada en la membrana induce la fusión entre la membrana plasmática y la envuelta viral, permitiendo de esta manera la entrada del virus en la célula (fig.4).

Fig. 4.Fases en la entrada del VIH en la célula. Papel de los correceptores.
Etapas tempranas. Retrotranscripción e integración viral
Una vez realizado el proceso de fusión entre las membranas viral y celular se produce la internalización de la nucleocápside viral y la desencapsidación del genoma viral. El proceso de síntesis de ADN a partir del ARN viral es realizado por el complejo enzimático de la transcriptasa inversa. Sin embargo, en un linfocito «en reposo», la retrotranscripción no finaliza la síntesis del ADN. Es necesario «activar» la célula infectada para que la retrotranscripción se complete. Si este fenómeno de activación no se produce, el ADN no integrado es degradado en un plazo corto (una o dos semanas) por las ADNasas celulares. Existe por tanto un compartimiento de ADN proviral no integrado que supone el 90% del existente en los linfocitos circulantes y que representa un reservorio susceptible de integración si la célula es adecuadamente activada. Debido a su corta vida media, la persistencia de ADN no integrado representa un marcador de replicación viral en pacientes en tratamiento antirretroviral, aunque éstos no presenten una carga viral detectable8.
Una vez sintetizado, el ADN proviral se acopla a una serie de factores celulares y virales formando lo que se denomina «complejo de preintegración», que es transportado al núcleo, donde el ADN viral se integra en el genoma del huésped constituyendo la forma proviral del VIH.
Etapas tardías: reactivación y replicación viral (fig. 1)
A partir del estado de integración, el VIH puede seguir un comportamiento variable: permanecer latente, replicarse de forma controlada o experimentar una replicación masiva con el consiguiente efecto citopático sobre la célula infectada9. Los linfocitos CD4 albergan mayoritariamente el genoma viral en forma latente. A partir del estado de provirus integrado, la replicación del VIH comienza mediante la transcripción del genoma viral. La parte inicial de este proceso, denominada «iniciación de la transcripción», depende de factores celulares y se produce en ausencia de proteínas virales. El principal factor celular implicado en el paso de la fase de latencia viral a la de reactivación es NF-kB, una familia de proteínas que regulan la transcripción de múltiples genes celulares implicados en los procesos de reconocimiento y activación inmunes. Este factor no existe en forma activa en los linfocitos CD4 en estado de reposo celular y es inducido únicamente en el curso de los procesos de activación inmunológica (fig. 5). Esto explica que la replicación del VIH dependa absolutamente de la activación de los linfocitos infectados. De esta manera, el linfocito CD4 representa un doble nicho ecológico en el ciclo biológico del VIH: en estado de reposo celular permite la latencia viral al carecer de los factores necesarios para permitir la replicación del VIH; por el contrario, la activación celular induce en el linfocito CD4 las proteínas de la familia NF-kB necesarias para iniciar la transcripción del genoma viral, transformándose así en una célula especialmente permisiva para la replicación del VIH. Este fenómeno de reactivación a partir del estado de latencia proviral es extraordinariamente rápido y agresivo y se ha estimado que, tras la activación linfocitaria, en 2 h se produce la síntesis de todas las proteínas virales en la célula, detectándose viriones viables entre 4 y 6 h después de la reactivación.
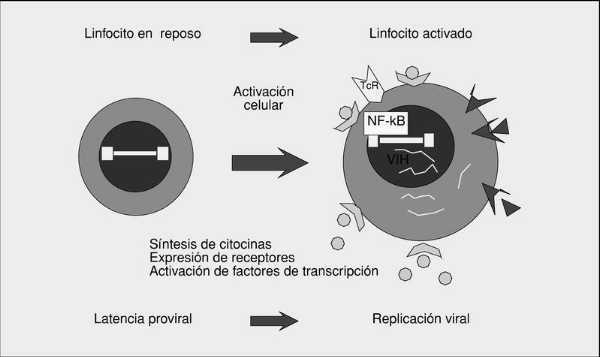
Fig. 5. Replicación del VIH y activación inmune.
El ARNm del VIH se sintetiza en forma de un único transcrito que debe ser transportado al citosol y procesado en ARN de distintos tamaños. Ambos procesos, procesamiento y transporte, son realizados fundamentalmente por otra proteína viral, Rev, que tiene una localización preferentemente nuclear. Después de su síntesis, las proteínas virales deben ser procesadas postraduccionalmente antes de ensamblarse en lo que constituirán las partículas virales maduras. En este proceso participan distintas proteínas virales entre las que destacan Vif, Vpu y la proteasa viral. El procesamiento de la gp160 en gp41 y gp120 se produce por una proteasa celular. La maduración final de los viriones y el ensamblaje correcto de las proteínas virales se produce en el momento final del ciclo infectivo, previamente a la gemación de los virus a través de la membrana celular, y permite constituir una partícula viral madura.
Ciclo de infección del VIH in vivo
La aplicación de las modernas técnicas de biología molecular, en especial las técnicas de amplificación genética e hibridación que detectan el ARN viral en plasma con alta sensibilidad y el estudio de los ganglios linfáticos de los sujetos infectados, ha permitido demostrar que la carga viral es detectable en todos los estadios de la enfermedad y que el VIH presenta una cinética de replicación elevada. La aplicación de modelos matemáticos ha demostrado que diariamente se producen en un sujeto infectado entre 109 a 1010 partículas virales, siendo la vida media de un virión de 0,3 días y la de un linfocito infectado que replica activamente el VIH, de 1,2 días. Estas cifras demuestran que el VIH presenta una cinética de replicación extremadamente agresiva, muy diferente a la de un lentivirus «clásico». Podemos dividir el ciclo viral in vivo en tres etapas: etapas tempranas de la infección, establecimiento de una infección crónica y fase acelerada.
Etapas tempranas de la infección
En la transmisión por vía sexual, las primeras células diana del virus son las células dendríticas y de Langerhans situadas en la submucosa y los linfocitos circundantes. En esta etapa se observa una transmisión prácticamente exclusiva de variantes con tropismo R5 (fig. 6). Se ha demostrado que las células dendríticas y de Langerhans producen la quimiocina SDF-1, por lo que CXCR4 se encuentra regulado negativamente en las células presentadoras y probablemente en los linfocitos asociados. Por el contrario, tanto las células de Langerhans como los linfocitos activados presentan el receptor CCR5 en la superficie, lo que explicaría la baja transmisibilidad de las variantes X4 y la selección de cepas con tropismo R5 en los estadios iniciales de la infección.
Una vez producida esta infección de la «primera estación» en la submucosa, la diseminación viral es inmediata y explosiva. Probablemente, la migración de células presentadoras y linfocitos, primero a los ganglios regionales y posteriormente a órganos linfoides distantes, sea el mecanismo por el que el virus se disemina en apenas 48-72 h, tiempo en el que se detecta ya un pool de linfocitos infectados similar al observado en la fase crónica de la infección. Se establece de esta manera muy rápidamente una infección crónica en la que el virus se acantona principalmente en los órganos lin foides11.
Fase crónica de la infección
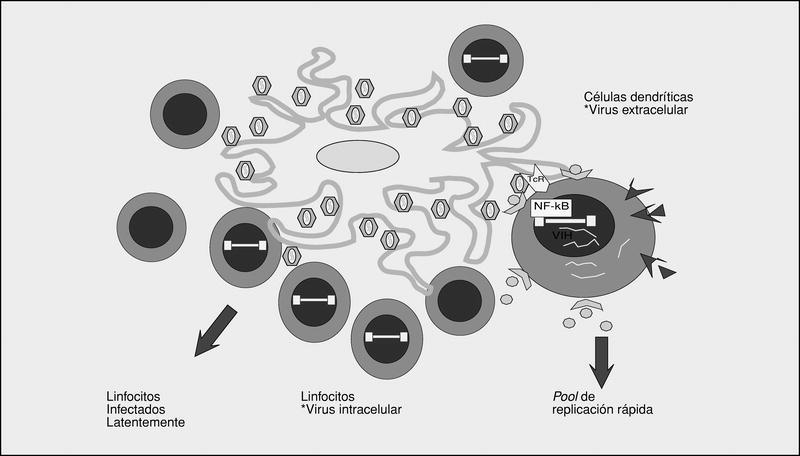
Los órganos linfoides representan el gran reservorio donde se producen los fenómenos de infección y propagación del VIH. En el estudio de los ganglios linfáticos (fig. 7) se observa una enorme cantidad de viriones extracelulares que se disponen esencialmente en los espacios interdigitantes de las células dendríticas, en estrecho contacto con los linfocitos. Esta acumulación de viriones en su membrana es debida a la interacción con una lectina recientemente caracterizada y que potencia la infectividad del VIH cuando las partículas virales interaccionan con los linfocitos en la superficie de las células dendríticas. Por otra parte, el VIH se detecta de forma intracelular en los linfocitos CD4, en la mayoría de modo latente. La población de linfocitos CD4 activados sería la responsable de la ingente producción de viriones observada en el paciente infectado y representaría la población destruida por efecto citopático directo que tendría una vida media inferior a 24 h. La progenie viral producida infectaría, a su vez, los linfocitos activados, especialmente aquellos que se encuentran en estadio de división para «regenerar» loslinfocitos destruidos. Se configura así un cuadro (fig. 8) en el que los centros germinales, en especial las prolongaciones interdigitantes de las células foliculares dendríticas recubiertas de virus, representan una interfase que facilita el reclutamiento y la infección de linfocitos activados que serán infectados productivamente por el VIH y destruidos por efecto citopático. Simultáneamente, existe un compartimientolinfocitario mayoritario en el que el virus permanece en un estado de latencia absoluta, sin que se conozca en la actualidad con precisión la cinética de transición de estas células latentes al compartimiento de replicación activa.
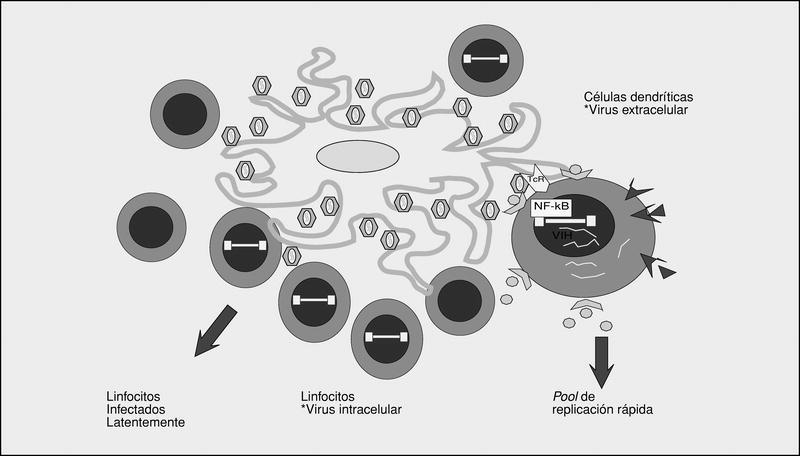
Fig 7. Infección en los centros germinales.

Fig 8. Cinética de replicación del VIH en órganos linfoides.
Estadio de sida
A medida que la infección progresa, la destrucción del sistema inmune tiene como consecuencia un aumento en la replicación viral. Esta replicación acelerada permite una mayor generación de mutantes, con lo que la posibilidad de evasión viral y la generación de variantes más citopáticas aumenta. La elevación de la carga viral y el rápido descenso en la cifra de linfocitos CD4 son los marcadores de una replicación «salvaje» del virus en ausencia de mecanismos de control inmunológico.
En esta fase se ha descrito la emergencia de variantes con un tropismo X4 y la detección de estas cepas se asocia con un rápido descenso en la cifra de linfocitos CD4 y un mal pronóstico de supervivencia. Algunos autores han planteado la posibilidad de que la envuelta de los virus X4 sea más «tóxica» que la de R5. Los mecanismos por los que estas variantes X4 emergen en los estadios finales sigue siendo un misterio. Es posible que la síntesis de SDF-1 en los órganos linfoides proteja a los linfocitos activados de la infección por variantes que utilicen CXCR4 como correceptor al bloquear el mismo y disminuir su expresión en la membrana. Según esta hipótesis, en el curso del proceso de destrucción y desestructuración del sistema linfoide serían destruidas las células productoras de SDF-1, facilitándose así la emergencia de variantes con tropismo X4 al disminuir los valores tisulares de SDF-1 (fig. 6).

Fig. 6. Evolución del tropismo del VIH a lo largo de la infección.
Mecanismos de inmunosupresión y destrucción lifocitaria (tabla 3)

Destrucción de CD4 por efecto citopático
La destrucción de los linfocitos CD4 representa el episodio más característico de la infección por el VIH. Dada la agresiva cinética de replicación viral, se postuló inicialmente que la destrucción de los linfocitos CD4 era una consecuencia directa de la replicación viral y del consiguiente efecto citopático en la célula infectada. Sin embargo, datos recientes demuestran que otros mecanismos de destrucción indirecta o de bloqueo linfo citario se hallan también implicados como causa del proceso de inmunosupresión, ya que la destrucción linfocitaria por efecto citopático directo no explica todos los fenómenos de disregulación inmune que se observan en el sida12.
Mecanismos indirectos de destrucción de CD4
Destrucción por mecanismos inmunes
En este grupo se engloban una serie de mecanismos por los que el propio sistema inmune originaría la destrucción de los linfocitos CD4 debido a un reconocimiento anormal o erróneo de sus dianas.
Reconocimiento de los CD4 infectados por linfocitos citotóxicos. Existen datos a favor de que este mecanismo pueda operar in vivo en la destrucción de CD4. En la fase de primoinfección existe una correlación entre el descenso de CD4 y la expansión de clones CD8 antivirales, y en experimentos in vivo se observa cómo la infusión de CD8 anti-VIH origina una disminución en el número de linfocitos CD4 infectados.
Por un fenómeno de «mirón inocente». Los linfocitos recubiertos de gp120 serían destruidos por el sistema inmunológico mediante reacciones de tipo ADCC (citotoxicidad celular dependiente del anticuerpo). Estos mecanismos se han documentado in vitro pero no hay evidencias claras de que existan in vivo.
Apoptosis
La apoptosis o muerte celular programada representa un mecanismo fisiológico mediante el cual la célula se «suicida» de forma controlada. Los mecanismos de apoptosis son naturales e incluso protectores frente al crecimiento celular incontrolado y cumplen un papel muy importante en todos los sistemas de desarrollo: embriogénesis, proliferación y diferenciación hematopoyética, control de la proliferación tumoral y regulación de la activación inmune. La apoptosis podría representar un mecanismo de destrucción de linfocitos CD4 en la infección por el VIH, que afectaría no sólo a las células infectadas sino que podría provocar la destrucción de linfocitos no infectados preactivados anormalmente por gp120 unida a sus receptores13. Se ha demostrado que en ganglios linfáticos de pacientes infectados existe in vivo una mayoría de células apoptóticas que no se encuentran infectadas y una minoría de células que replican activamente el virus y no presentan signos de apoptosis. Estos datos sugieren que la apoptosis puede ser un mecanismo indirecto complementario de la destrucción por efecto citopático directo.
Bloqueo de la activación y proliferación de CD4
Los modelos de replicación viral y destrucción linfocitaria por efecto citopático directo estiman que diariamente alrededor de 108 linfocitos son destruidos. Esto implica que una cantidad similar de células debe ser producida diariamente para mantener el número de CD4 y alcanzar un estado de equilibrio en que el número de células destruidas sea compensado por un estado anormal de proliferación CD4 que sometería al sistema a un estrés inmunológico muy importante. Sin embargo, cuando se ha podido medir la cinética de recuperación de CD4 tras el tratamiento antirretroviral de alta eficacia, se observa un aumento en la cinética de división linfocitaria en la subpoblación CD4 respecto a los sujetos infectados sin tratamiento y a los pacientes seronegativos. Estos datos demuestran que durante la fase crónica de la infección no sólo no existe un turnover aumentado de CD4 sino que, por el contrario, existe un bloqueo de la entrada de los linfocitos CD4 en ciclo de división14. Únicamente con la disminución de la carga viral plasmática a valores indetectables el sistema es capaz de entrar en ciclo de mitosis con una mayor velocidad. Estos datos apuntan a un nuevo mecanis mo de disminución de CD4: el bloqueo en la activación y proliferación linfocitaria debido a la infección por el VIH. Este bloqueo puede producirse en los sistemas central (timo o médula ósea) o periférico (órganos linfoides).
Respuesta inmune frente a la infección por el VIH
En el paciente infectado por el VIH se ha descrito una respuesta inmune intensa que abarca prácticamente todos los mecanismos efectores del sistema inmunitario (tabla 4). Esta respuesta es, asimismo, relativamente amplia, ya que se desarrolla frente a numerosos epítopos y prácticamente todas las proteínas del virus, tanto estructurales como reguladoras, son reconocidas como extrañas.

Respuesta humoral
Inmunidad específica
La infección por el VIH induce una intensa respuesta de anticuerpos frente a prácticamente todas las proteínas reguladoras y estructurales del VIH. Algunos de los anticuerpos generados, especialmente los dirigidos frente a gp41 y frente a los dominios variable 3 (V3) y de interacción con CD4 de la proteína gp120, tienen carácter neutralizante in vitro y en experimentos de inmunoterapia adoptiva in vivo, por lo que se postula la acción protectora de dichos anticuerpos15. Sin embargo, in vivo la producción de anticuerpos con capacidad neutralizante es muy escasa. Esto es probablemente debido a que desde el punto de vista estructural, el dominio de neutralización se encuentra oculto en la conformación nativa de la gp160 y sólo tras el desplegamiento de la misma se expone el dominio V3 y regiones adyacentes que interaccionarán con los correceptores de quimiocinas. Por tanto, las partes expuestas y más inmunógenas de la proteína en su forma «compacta» inducen la síntesis de anticuerpos no neutralizantes, ya que no son epítopos de interacción con el receptor, mientras que sólo cuando la proteína se despliega por la unión a CD4 se exponen epítopos capaces de inducir anticuerpos neutralizantes de amplio espectro (epítopos inducidos por la unión a CD4).
Se ha descrito que el suero de pacientes seropositivos puede presentar un efecto «facilitador» de la infección por el VIH. Este fenómeno se debería a la presencia de anticuerpos dirigidos frente a la envoltura del virus, que opsonizarían las partículas virales y las harían susceptibles de ser fagocitadas por los monocitos-macrófagos, con lo que paradójicamente aumentarían la infección de éstos por el VIH. La importancia de este fenómeno in vivo es desconocida.
Inmunidad inespecífica
Existen numerosos factores solubles que son activos frente a la infección por el VIH. Los virus en general y el VIH en concreto son sensibles a la inhibición por complemento. Los interferones también tienen actividad antiviral frente al VIH. Probablemente todos estos mecanismos de inmunidad humoral inespecífica representen una barrera frente a la infección por el VIH, pero son insuficientes para permitir un control completo de la replicación viral.
Respuesta celular
Inmunidad específica
El estudio de la respuesta citotóxica in vitro (CTL) ha demostrado que en los pacientes seropositivos existe una expansión clonal de linfocitos CD8 con actividad citotóxica16. Esta respuesta celular es particularmente intensa en pacientes en estadio de primoinfección y en algunos pacientes con reconstitución inmune obtenida tras tratamiento antirretroviral. Desde el punto de vista cualitativo, esta respuesta es completa en un doble sentido: se reconocen múltiples epítopos de las distintas proteínas del virus, y el estudio del repertorio de utilización de receptores antigénicos de linfocitos T revela que se utiliza un amplio espectro de reordenamientos. No parece que existan en la respuesta celular específica agujeros en el repertorio o una deleción clonal de determinados Vß. Sin embargo, en pacientes infectados estables que presentan un deterioro en su evolución se ha observado un escape viral en relación con la generación de variantes virales incapaces de ser reconocidas por el repertorio linfocitario T previamente establecido. La respuesta celular específica no se limita a los linfocitos CD8; se ha demostrado una respuesta CD4 específica que probablemente sea importante para la puesta en marcha de una respuesta inmune eficaz frente al VIH. El problema de estos estudios es que sólo permiten establecer correlaciones, pero es difícil concluir una relación causa-efecto entre la aparición de un tipo específico de respuesta inmune y el control de la replicación viral. Aun teniendo en cuenta estas limitaciones, los datos actualmente comunicados sugieren que la respuesta celular helper y citotóxica pueden ser importantes para contener la replicación viral en estadios precoces de la enfermedad en que existe una indemnidad relativa del sistema inmune.
Inmunidad inespecífica
En cuanto a los mecanismos de inmunidad celular no específicos (no restringida por el sistema HLA) se ha descrito la existencia de citotoxicidad celular dependiente de anticuerpos (ADCC), así como una actividad citotóxica natural (NK) frente al VIH en pacientes seropositivos. Tanto en la respuesta celular específica (CTL) como en la no restringida por el sistema HLA (ADCC y NK), la actividad antiviral es más intensa en los estadios asintomáticos de la infección y se ha postulado que el mantenimiento en niveles elevados de la respuesta citotóxica sería un factor de buen pronóstico en la evolución a sida. Si estas modificaciones son causa o efecto de la progresión a sida no ha sido claramente demostrado.
Factores solubles
Desde hace años se sabe que los linfocitos CD8 de pacientes seropositivos inhiben la replicación del VIH y que esta actividad supresora corresponde a factores solubles presentes en el sobrenadante generado a partir de linfocitos CD8 activados. La descripción de que los receptores frente a los quimiocinas son, a su vez, correceptores frente a distintas cepas del VIH ha permitido caracterizar que esta actividad supresora corresponde a quimiocinas como RANTES, MIP-1*, MIP-1ß y SDF-1.
Respuesta inmune en los distintos estadios de la infección (fig. 9)

Fig 9. Evolución de los parámetros virológicos e inmunológicos en la infección por el VIH.
Primoinfección
Tras el contacto con el VIH se produce un período ventana de 4-12 semanas, que corresponde a la fase de primoinfección y durante el cual no es posible detectar la presencia de anticuerpos específicos frente al VIH, a pesar de existir grados de viremia muy elevados. Durante este período es posible detectar actividad citotóxica frente al VIH; la existencia de esta actividad celular en ausencia de anticuerpos sugiere que la respuesta celular es más precoz e importante en el control inicial de la replicación viral que la síntesis de anticuerpos. Probablemente, ambos brazos de la inmunidad humoral y celular son importantes en el control de la replicación viral tras la primoinfección. Este control es el resultado del equilibrio entre dos factores: la virulencia de las cepas infectantes y la intensidad de la respuesta antiviral generada por el huésped. La resultante de estos dos factores se refleja en la carga viral basal del paciente tras la primoinfección, que representa un dato de enorme valor pronóstico en la evolución de la infección, ya que indica el equilibrio alcanzado en un sujeto determinado entre el virus y su sistema inmune17. En cualquier caso, esta respuesta antiviral es incapaz de erradicar el virus que ya se ha acantonado en las primeras horas de la infección en el organismo y se limita a contener aunque sea en una proporción importante la replicación viral. Se establece así una infección crónica persistente en el sujeto infectado.
Fase crónica de la infección
En la fase crónica de la enfermedad se mantienen durante años respuestas celulares y humorales intensas frente al VIH. Esta falta de atenuación de la respuesta refleja, por una parte, la intensidad y la cronicidad de la replicación viral que sigue estimulando persistentemente el sistema inmune y, por otra, la capacidad de éste para controlar durante largos períodos la replicación masiva que se produce a lo largo de toda la enfermedad. Sin embargo, los mecanismos de inmunosupresión y de destrucción de los linfocitos CD4 por el VIH se producen de forma persistente, y a medio plazo conllevarán una incapacidad progresiva del sistema inmune para contener la replicación viral. A esto se unirá la emergencia de variantes más agresivas que aumentarán la destrucción inmunológica y desplazará el equilibrio virus-hospedador a una situacion de replicación viral acelerada y de profunda inmunosupresión.
Estadio avanzado de la enfermedad
Los estadios finales de la enfermedad se caracterizan clínicamente por la aparición de infecciones oportunistas, desde el punto de vista inmunológico por la disminución del número de linfocitos CD4 y virológicamente por la elevación de la carga viral. En esta etapa se observa un deterioro de la respuesta humoral y celular frente al VIH: disminuyen los valores de anticuerpos frente a p24 y otras proteínas virales, decrece la tasa de anticuerpos neutralizantes, la actividad citotóxica y el número de linfocitos CD8, y se observa un deterioro en la actividad ADCC y NK. Probablemente, este deterioro refleja la destrucción masiva del sistema inmune por una replicación viral acelerada que permite la generación de mutantes de escape, incapaces de ser contenidas por el sistema. Se entra así en un círculo vicioso en el que el deterioro inmunológico progresivo permite una replicación viral más agresiva.
Respuesta inmune en los pacientes no progresores
Se ha descrito que en los pacientes con evolución lenta a sida y un número estable de linfocitos CD4 la función inmune se encuentra bien conservada. En estos sujetos los mecanismos efectores frente al VIH presentan una actividad muy superior a la encontrada en los pacientes que tienen una evolución rápida a sida. Estas diferencias cuantitativas pueden reflejar simplemente la existencia de una replicación viral menos agresiva. De hecho, en algunos pacientes se ha descrito la infección por variantes virales profundamente defectivas. Por otra parte, se ha descrito que un porcentaje elevado de los pacientes con evolución lenta de la infección presenta defectos en los correceptores del VIH asociados con una resistencia a la infección y a la progresión de la enfermedad. Probablemente, los progresores lentos representan un grupo heterogéneo de pacientes con distintos mecanismos de protección frente a una evolución rápida o habitual de la infección por el VIH: factores genéticos, infección por variantes defectivas o una respuesta inmune especialmente eficaz18.
Mecanismos de protección en sujetos expuestos al virus y no infectados (tabla 5)

Uno de los aspectos más llamativos en el estudio de la infección por el VIH es la existencia de sujetos que, a pesar de numerosas exposiciones al VIH, no se infectan. Este grupo de sujetos, denominados «expuestos no infectados» (ENI), ha sido objeto de un estudio intenso, ya que la definición de los mecanismos de protección en los mismos reviste gran interés para la comprensión de los mecanismos inmunes de protección frente al VIH in vivo. En ellos se ha definido una serie de factores genéticos «protectores» frente a la infección, como un haplotipo HLA particular (B35) o la homozigosis para la deleción 32-CCR5. Otros autores han demostrado que en algunos sujetos ENI existe una memoria inmune frente al VIH en distintas formas (producción de interleucina 2, actividad proliferativa en respuesta a péptidos del VIH, altos valores de IgA secretora en vagina19), que sugieren la existencia de un contacto previo con el virus y una sensibilización del sistema inmune frente al virus, con la generación de una respuesta antiviral específica en sujetos no infectados. Por último, también se ha descrito que en determinados casos, las cepas a las que los sujetos ENI se ven expuestos se caracterizan por su bajo grado de replicación en cultivo o por características cuantitativas (baja carga viral) o cualitativas (tropismo) del virus que indican una baja agresividad20. No puede excluirse una combinación de todos estos factores y es posible que en determinadas situaciones la baja carga viral con la que se entra en contacto o las características cualitativas del virus que impiden una buena propagación de la infección induzcan una respuesta inmune protectora en el hospedador. En estos casos existiría una «vacunación» natural con un virus defectivo o en una dosis insuficiente para infectar, pero suficiente para inmunizar al sujeto.
Mecanismos de escape viral
Variabilidad genética
La tasa de variabilidad del VIH es similar a la de otros virus ARN debido a que la transcriptasa inversa tiene una tasa de error de 10-3-10-4 por nucleótido y sonda de copia. Esta falta de fidelidad tiene una doble consecuencia: por una parte, se produce una gran proporción de virus defectivos y por otra, se genera una alta diversidad en las proteínas del virus que le permiten escapar al control de la respuesta inmune específica. El VIH dispondría de un mecanismo clásico de escape inmune frente al VIH común a los virus ARN, en los que el alto índice de variabilidad les permite encontrar agujeros en el repertorio inmunológico.
Enmascaramiento de epítopos de neutralización
La estructura del envoltorio viral en su forma nativa oculta los epítopos susceptibles de neutralización por anticuerpos, lo que explicaría la baja tasa de anticuerpos neutralizantes generados por los pacientes seropositivos. Por otra parte, al producirse estos cambios en el momento de interacción de las membranas viral y celular, la eficacia de los anticuerpos es muy reducida debido a la baja accesibilidad de los mismos.
Latencia y reactivación
Una célula latentemente infectada escapa de manera absoluta a la vigilancia imunológica al no expresar productos virales en la membrana de la célula infectada. La reactivación del VIH a partir de su estado de latencia es un proceso rápido y masivo, por lo que la generación de nuevos viriones se produce antes de que la célula sea destruida por el sistema inmune. Por otra parte, estos procesos de reactivación-reinfección se producen en los centros germinales de los órganos linfoides a los que llegan mal los anticuerpos y que presentan un microambiente celular idóneo para el proceso de infección: las células dendríticas cuya membrana se encuentra tapizada por viriones interacciona con los linfocitos y facilitan su infección. La activación linfocitaria en este microambiente, a su vez, aumenta la infectividad de las células diana y la replicación viral.
Infección de reservorios
Se ha demostrado que el VIH infecta in vivo células de estirpe mononuclear-fagocítica, como la microglía cerebral, las células de Langerhans y las células dendríticas de localización submucosa. Las células infectadas en el SNC probablemente sean importantes no sólo en relación con la lesión neurológica sino también como reservorio de la infección. Debido a la baja permeabilidad de la barrera hematoencefálica a determinados fármacos y el carácter de «santuario inmunológico» del SNC, éste podría constituir un reservorio de gran importancia en cuanto a replicación viral residual, diversificación antigénica y generación de variantes resistentes a fármacos. Es posible que otros compartimientos celulares como el sistema reproductor o especialmente los órganos linfoides sean un reservorio de la infección debido a una baja biodisponibilidad farmacológica o a la escasa accesibilidad de los anticuerpos. La existencia de una replicación persistente en esta localización a
pesar de la terapia antirretroviral apoyaría esta hipótesis y la posibilidad de que el VIH se acantone en distintos reservorios en los que se replicaría activamente y se propagaría a pesar de un tratamiento aparentemente eficaz.
Algunos interrogantes planteados en el momento actual
¿Cuáles son los mecanismos inmunes eficaces frente al VIH?
Esta cuestión tiene un gran interés para el diseño de vacunas terapéuticas y preventivas y estrategias de inmunoestimulación, así como para poder evaluar la eficacia de intervenciones cuyo objetivo es potenciar la respuesta inmune con el fin de conseguir un control del VIH. Los anticuerpos probablemente tengan un papel muy limitado en el control de la infección por el VIH ya que, a pesar de que existen valores elevados de anticuerpos frente a prácticamente todas las proteínas del VIH, las tasas de anticuerpos neutralizantes son muy bajas. Las respuestas celulares parecen mucho más eficaces en el control de la replicación viral. De hecho, este tipo de respuesta es muy intenso en dos tipos de situaciones: en los pacientes supervivientes a largo plazo y durante la primoinfección, especialmente si el paciente es tratado tempranamente.
¿Cuáles son los mecanismos de protección frente a la transmisión del VIH por vía sexual?
Una de las preguntas más importantes planteadas en la historia natural de la infección por el VIH radica en la comprensión de los mecanismos implicados en la protección de sujetos que a pesar de tener un contacto repetido y estrecho con el virus no se infectan. Trabajos recientes demuestran que tanto las características de las cepas virales del sujeto infectado como la inmunidad local generada serían elementos implicados en la aparente resistencia a la infección. Probablemente exista una combinación de ambos factores: un inóculo viral insuficiente para producir la infección pero capaz de inducir una respuesta inmune de forma local y sistémica que actúe como una inmunización natural del sujeto expuesto y no infectado.
¿Cuál es el papel de las quimiocinas y sus receptores en la protección frente a la infección viral y en el control de la progresión de la infección?
Los datos actuales demuestran que los sujetos homozigotos para una deleción en el dominio aminoterminal del receptor CCR5 son resistentes a la infección por el VIH in vitro y aparentemente in vivo por cepas de tipo R5. El rasgo heterozigoto en CCR5 protegería parcialmente frente a la progresión, pero no frente a la infección. Recientemente se han descrito nuevas mutaciones en CCR5 y también en CCR2, CX3CR1 y SDF-1 que confieren una resistencia a la infección y aceleran o enlentecen la progresión de la enfermedad. Sin embargo, persiste una serie de preguntas. ¿Por qué los sujetos defectivos en el receptor CCR5 se infectan en tan baja proporción con cepas CXCR4 trópicas para las que disponen de receptores? ¿Por qué se seleccionan en los estadios iniciales de la infección cepas de tipo R5 y las cepas de tipo X4 sólo se detectan en los estadios finales de la enfermedad? ¿Son las quimiocinas factores «naturales» de protección que explicarían la distinta rapidez de progresión a sida en los distintos sujetos?
¿Hasta qué punto se produce la regeneración del sistema inmune con el tratamiento antirretroviral?
En las primeras semanas de tratamiento se observa un aumento de las células memoria en sangre periférica, debido a una redistribución de órganos linfoides a sangre periféricapero no un aumento en el número absoluto de linfocitos naïf21. Desde el punto de vista clínico, en este período no existe una protección absoluta frente a determinadas infecciones oportunistas al ser una reconstitución aparente y no real (tabla 6). En una segunda etapa se produce un aumento en el número de linfocitos naïf y en muchos pacientes un aumento en los tests de respuesta a antígenos in vitro que se asocia desde el punto de vista clínico con un control de las infecciones oportunistas. El grado de reconstitución inmune guarda una relación directa con el deterioro inmunológico previo al tratamiento: aquellos pacientes con unos valores elevados de CD4 (> 500 células/µl) recuperan cifras normales o casi normales de CD4, así como una respuesta funcional frente a antígenos y mitógenos in vitro. Por el contrario, una proporción de pacientes con cifras muy disminuidas de CD4 antes de empezar tratamiento antirretroviral no recuperan valores suficientes de linfocitos y la respuesta en los tests de transformación linfoblástica a distintos antígenos es irregular22.

La reconstitución del sistema inmune es secundaria a la expansión de linfocitos CD4 no sólo en el sistema periférico, sino también en el central. Uno de los hallazgos más sorprendentes del estudio de la reconstitución inmunológica tras el tratamiento antirretroviral es la detección de células generadas en el timo23, lo que ha obligado a replantear el papel de este órgano en la vida adulta. Como se ha mencionado, la infección por el VIH no sólo produce una destrucción directa de linfocitos CD4, sino un bloqueo en la producción de los mismos. El control de la replicación viral neutraliza ese bloqueo permitiendo la generación de poblaciones linfoides jóvenes en el timo, lo que abre una nueva perspectiva en el alcance de la reconstitución inmune en los pacientes con buena respuesta al tratamiento.
¿Puede el sistema inmune reconstituido contribuir a la erradicación de la infección por el VIH?
La demostración de que la reconstitución inmune es posible tras el control de la replicación del VIH y que en algunos casos esta recuperación inmunológica lleva aparejada un aumento en los grados de actividad anti-VIH plantea la posibilidad de que el propio sistema inmune sea el elemento decisivo para reducir la viremia a valores que permitan la supresión de la medicación y una supervivencia prolongada24. El objetivo planteado actualmente no es por tanto la erradicación completa mediante medicación, sino la potenciación de la respuesta inmune por distintos medios para transformar a los pacientes en «supervivientes a largo plazo». La gran pregunta planteada en el contexto de la erradicación es si la reconstitución inmunológica permitirá una mejor respuesta frente al VIH, y si no es así ¿por qué no funciona?
¿Cuáles son las estrategias de potenciación de la respuesta inmune frente al VIH en combinación con la terapia antirretroviral?
Tratamiento con interleucina 2
La utilización de interleucina 2 (IL2) se realiza con un doble objetivo: aumentar los valores de linfocitos CD4 e inducir la reactivación de los reservorios latentes para aumentar la eficacia de la medicación antirretroviral. Sin embargo, hasta el momento no se ha demostrado un beneficio claro del empleo de IL2 en el tratamiento de la infección por el VIH.
Las interrupciones controladas del tratamiento
Existen casos aislados de pacientes que tras la suspensión de tratamiento realizan repuntes de carga viral a cifras muy inferiores a sus valores basales, y en algunos casos no presentan carga viral detectable. Estos hallazgos han llevado a plantear estudios reglados de interrupciones estructuradas de tratamiento (STI). Los resultados publicados hasta el momento no son muy alentadores, ya que se observa en la mayoría de pacientes un repunte brusco de la carga viral que alcanza los valores basales del paciente en un período máximo de 4-6 semanas25.
Los ensayos de interrupción de tratamiento tienen un interés indudable pero todavía no han demostrado su eficacia. Dado el riesgo potencial que suponen para el paciente, deben realizarse de forma extremadamente controlada por equipos con el soporte clínico y de laboratorio requeridos para realizar este tipo de estudios. Probablemente, en el próximo año se obtengan datos concluyentes sobre las situaciones en que estos ensayos pueden tener una utilidad en el control de la replicación viral por el sistema inmune en ausencia de tratamiento antirretroviral.
Utilizacion de vacunas terapéuticas. ¿Qué posibilidades tienen las estrategias de vacunación terapéutica en el momento actual?
La supresión de la viremia y la reconstitución inmune suponen un nuevo contexto en el que la inmunización puede tener un beneficio potencial si consigue incrementar la memoria inmune frente al VIH en los pacientes vacunados.
Los datos publicados hasta el momento ofrecen resultados contradictorios: en algunos estudios no se observa un efecto positivo, mientras que en otros trabajos se detecta un incremento en parámetros de respuesta específica frente al VIH: inducción de CTL, síntesis de citocinas en respuesta a antígenos virales in vitro y, en algunos ensayos, disminución de carga viral. Sin embargo, prácticamente la totalidad de los trabajos presentados son estudios abiertos con una gran heterogeneidad en cuanto al antígeno utilizado, las características de los pacientes y las condiciones de inmunización. Únicamente estudios controlados a doble ciego a largo plazo permitirán evaluar la mejora en la respuesta inmune inducida por las vacunas terapéuticas y si estos cambios inmunológicos se traducen o no en un beneficio clínico para los pacientes.
¿Qué características debe reunir una vacuna preventiva frente al VIH?
Una vacuna frente al VIH debe ser capaz de inducir una respuesta inmunitaria celular y probablemente humoral intensa, no sólo sistémica sino muy especialmente en las mucosas, con el fin de conferir una buena protección frente a la infección por vía sexual. Asimismo, debe ser capaz de inducir memoria inmunológica prolongada, que es una de las limitaciones más importantes de los distintos modelos de vacunas experimentales generadas hasta la actualidad. Los desarrollos actuales en vacunas se basan principalmente en el modelo VIS/macaco infectados por virus seudotipados con la envuelta del VIH (SHIV) sistemas de inmunización que se basan en la utilización de vectores derivados de poxvirus que contienen fragmentos de distintos genes del VIH (gag, pol, env). Las vacunas de virus atenuados se encuentran prácticamente desechadas. Aunque en macacos adultos se ha demostrado el papel protector de virus defectivos (delecionados en el gen Nef), la inducción de enfermedad observada en neonatos y también en algunos macacos adultos a largo plazo con estas preparaciones vacunales y los problemas éticos que plantea la inmunización con retrovirus «atenuados» hacen de este planteamiento algo inviable por el momento. El dato más esperanzador del que se dispone en el terreno de las vacunas reside en la observación de que la respuesta inmune generada tras la primoinfección es muy eficaz en el control de la infección aunque no consiga su erradicación. Es previsible que una inmunización previa a la infección con vectores suficientemente inmunógenos pueda inducir una respuesta protectora frente a la infección.
Conclusión
En los últimos años se han producido avances de gran importancia en la comprensión de la inmunopatología de la infección por el VIH. Puede decirse que la inmunología del sida se desarrolla por fin sobre bases científicas sólidas que han sido posibles gracias a dos grandes hitos: la caracterización de los correceptores del VIH y el estudio de la reconstitución inmune tras el tratamiento antirretroviral. Estos descubrimientos nos han permiti-do caracterizar el tropismo viral, identificar mecanismos genéticos de protección frente a la infección, comprender los mecanismos de destrucción linfocitaria y aquellos aspectos de la respuesta inmune que son eficaces en el control de la replicación del VIH. Aunque persisten muchas incógnitas, y a pesar de su enorme complejidad, la comprensión de los mecanismos inmunológicos de protección frente a la infección y la fisiopatología de la inmunodeficiencia producida por el VIH siguen siendo eslabones esenciales para conseguir un tratamiento preventivo y curativo de esta enfermedad.