








En las últimas décadas, los progresos alcanzados en el campo de la genética, la biología molecular, la fisiología, la bioquímica y la neuroimagen de la enfermedad de Parkinson (EP) han mejorado el conocimiento de la enfermedad y han permitido un mejor diagnóstico y tratamiento de los pacientes. En correlación con los estudios clinicopatológicos realizados en enfermos con EP, se han propuesto diversos criterios clínicos con el objetivo de mejorar la precisión diagnóstica y evitar, en lo posible, errores en los estudios clínicos y epidemiológicos que se realizan. Por otra parte, el hallazgo de nuevas causas genéticas de algunos casos familiares de EP ha supuesto un importante adelanto en el conocimiento de la etiología de la enfermedad. Por último, el desarrollo de nuevos fármacos y la aplicación de la neurocirugía funcional han supuesto una mejoría significativa en el tratamiento de los síntomas y de las complicaciones inducidas por el tratamiento crónico con l-DOPA.
La enfermedad de Parkinson (EP) se caracteriza por un síndrome clínico que cursa con temblor, acinesia, rigidez e inestabilidad postural, que obedece a una patología subyacente específica, definida por una pérdida masiva de neuronas dopaminérgicas pigmentadas de la sustancia negra (SN) con el depósito de cuerpos de Lewy. Se considera que la presencia de 2 de los 3 signos motores cardinales (temblor, rigidez, bradicinesia) y una respuesta favorable y mantenida a la l-DOPA (70-100%) son esenciales para el diagnóstico de EP. Además, la EP también se define por la ausencia de otras causas de parkinsonismo1.
Es importante que el clínico sea capaz de diferenciar la EP de otras formas menos comunes de parkinsonismo, dado que el pronóstico y el tratamiento pueden diferir considerablemente.
Manifestaciones clínicas
Las principales manifestaciones clínicas de la EP se reflejan en la tabla 1.

Inicio de la enfermedad
Se cree que la pérdida de neuronas nigrales dopaminérgicas empieza varios años antes del inicio de la clínica y los síntomas aparecen cuando un 80% de las neuronas dopaminérgicas nigrales han degenerado2. El inicio de la enfermedad es lentamente progresivo, y con pródromos de síntomas inespecíficos3. Fatiga, sensación de malestar indefinida o cambios de carácter pueden aparecer años antes de los primeros síntomas motores. Los síntomas motores son: sensación de disminución de fuerza, incoordinación o dificultad para escribir. El dolor o tensión en la musculatura del hombro o de un brazo es un síntoma inicial frecuente. Un examen neurológico meticuloso puede revelar, en estadios iniciales, signos de parkinsonismo subclínico (p. ej., temblor intermitente de uno o varios dedos de la mano, asimetría en el tono muscular de las extremidades superiores o rigidez en rueda dentada).
Signos motores
Temblor
El temblor de reposo es la manifestación inicial más frecuente. Se presenta en un 79-90% de los pacientes4,5 y en un 76-100% en series autópsicas con diagnóstico de EP confirmado6,7. Tiene una frecuencia de 3-6 Hz, se inicia distalmente en una extremidad, generalmente en una mano, y en menor frecuencia en los labios, mandíbula, lengua o en una extremidad inferior. Desaparece durante el movimiento, para reaparecer cuando la extremidad vuelve al reposo después de un breve intervalo de tiempo sin temblor. El clásico temblor de «contar monedas» suele desaparecer cuando el temblor avanza y se extiende al brazo. Con el tiempo, suele extenderse a la pierna del mismo lado y, por último, a las extremidades contralaterales. Aunque en la enfermedad avanzada el temblor es bilateral, normalmente se mantiene asimétrico durante el curso de la enfermedad. A menudo el temblor de las manos aumenta al caminar, por lo que puede observarse cuando se examina la marcha del paciente. El temblor parkinsoniano desaparece durante el sueño y empeora con el cansancio, el estrés y con la ansiedad.
Rigidez
La rigidez, que está presente en más del 90% de los pacientes con EP, es causada por un incremento del tono muscular que afecta todos los grupos musculares (flexores, extensores, músculos axiales o de extremidades). La rigidez se manifiesta como un aumento de resistencia al movimiento pasivo de una articulación. En la EP, esta resistencia se percibe a lo largo de todo el movimiento. En muchos pacientes puede encontrarse una rigidez en rueda dentada que refleja un temblor subyacente. Este aumento de tono puede reforzarse cuando el paciente efectúa movimientos voluntarios de la extremidad del otro lado (signo de Froment). Al igual que el temblor, la rigidez suele ser asimétrica a lo largo de la evolución de la enfermedad, aunque en estadios avanzados es casi siempre bilateral.
Acinesia
Es el signo más incapacitante de la EP. Prácticamente todos los pacientes presentan dificultad para efectuar movimientos secuenciales y simultáneos, y son muy sensibles a estímulos externos que pueden llegar a impedir el movimiento. Al principio afecta tan sólo a músculos distales (micrografia), pero con el tiempo se afectan todos los grupos musculares y causa múltiples dificultades en las actividades de la vida diaria. En la exploración de los movimientos repetitivos rápidos, como la oposición repetida de los dedos pulgar e índice, se observa una disminución de la amplitud y de la frecuencia del movimiento. Los pacientes suelen referir la acinesia como dificultad para levantarse de una silla, movilizarse en la cama y, en general, para todos los movimientos. La hipomimia, la hipofonía, la disartria y la sialorrea son manifestaciones de la acinesia en otras áreas musculares.
Alteración de la marcha e inestabilidad postural
Uno de los primeros signos de la EP es la disminución del braceo al caminar. Más tarde presentan enlentecimiento de la marcha, los pasos se hacen cortos, arrastrando las piernas. Más tarde aparece la «festinación», que es una marcha caracterizada por pasos progresivamente más rápidos y cortos, durante la cual el enfermo no puede detenerse voluntariamente. También son fenómenos avanzados de la EP los bloqueos o «congelación», especialmente durante el inicio de la marcha, con los giros, o al aproximarse a lugares estrechos8. La inestabilidad postural, junto con el trastorno de la marcha, es, normalmente, el último síntoma cardinal de la EP, y el que responde peor al tratamiento con l-DOPA. La pérdida de los reflejos posturales se caracteriza por la dificultad para corregir rápidamente el equilibrio frente a un empujón de intensidad variable.
Alteraciones oculares
Al contrario de lo que ocurre en muchos síndromes de parkinsonismo plus, la función oculomotora está preservada en la EP. Es frecuente la limitación de la mi rada vertical superior o la presencia de movimientos oculares sacádicos hipométricos, como también puede verse en ancianos sanos9. La de la mirada vertical es una presentación típica de la parálisis supranuclear progresiva (PSP). Los pacientes con EP pueden referir
visión borrosa, secundaria a alteraciones de la convergencia que puede ser debida a un efecto colateral del tratamiento con anticolinérgicos o antidepresivos tricíclicos. La frecuencia de parpadeo está claramente disminuida en la EP, lo que contribuye a la falta de expresión facial.
Alteraciones orofaríngeas
Los pacientes con EP presentan un habla hipocinética, monótona e hipofónica10. A veces el lenguaje se hace más lento al final de las frases o, contrariamente, es rápido (festinante). Con el avance de la enfermedad el lenguaje puede llegar a ser incomprensible. La sialorrea es consecuencia de la disminución del tránsito de la saliva a la faringe11. La disfagia, consecuencia de la acinesia faríngea, aparece en estadios avanzados y puede ser causa de aspiraciones pulmonares graves. Una disfagia grave al inicio de la clínica es más típica en otras formas de parkinsonismos atípicos como la PSP o la atrofia multisistémica (MSA). Otro síntoma es la disminución de la función olfatoria, que aunque precoz y frecuente, en general no es advertida por el paciente12.
Manifestaciones no motoras
Deterioro cognitivo
En los estadios iniciales de la enfermedad se han encontrado déficit cognitivos leves o moderados afectando la función visuoespacial, la atención, las funciones ejecutivas y la fluencia verbal13. Los pacientes pueden presentar un pensamiento lento, y responder lenta pero apropiadamente (bradifrenia). Los signos de demencia, si se presentan al inicio de la enfermedad, sugieren otras posibles etiologías distintas de la EP, como la enfermedad de Alzheimer (EA), la enfermedad por cuerpos de Lewy difusos (EDCL), la PSP o la enfermedad de Creutzfeldt-Jakob (ECJ). En estadios más avanzados de la enfermedad, la demencia puede afectar hasta un 20-30% de los pacientes14. La demencia de la EP sigue un patrón de afección predominantemente subcortical en contraposición a la demencia de tipo cortical, típica de la EA. Los signos de la demencia subcortical son bradifrenia, lentitud psicomotora, depresión, ausencia de afasia, y escasa afección de la memoria. En la práctica clínica la demencia en la EP es, en muchas ocasiones, indistinguible de la demencia de la EA. La demencia es un grave problema en la EP y es causa frecuente de ingreso del paciente en instituciones geriátricas.
Alteraciones psiquiátricas y del sueño
La depresión es el trastorno afectivo más frecuente en la EP y afecta a un 50% de los pacientes15 y en un 50% de los casos puede ser una manifestación inicial16, aunque sólo un 2-7% de los pacientes tienen criterios de depresión mayor17. En algunos pacientes es reactiva a la limitación motora de la EP. Responde al tratamiento antidepresivo convencional.
La ansiedad es frecuente18 en forma de crisis de angustia, sobre todo se da en los períodos off o de inmovilidad inducidos por la caída del efecto terapéutico de la l-DOPA tras años de tratamiento.
También son habituales los trastornos del sueño. Es frecuente la reducción en el sueño de ondas lentas, múltiples despertares y disminución del sueño REM19. En los pacientes tratados con l-DOPA, la presentación de sueños vívidos y pesadillas en ocasiones pueden preceder durante semanas o meses a una psicosis dopaminérgica.
Disautonomía
La presentación precoz de signos y síntomas disautonómicos es un indicador de parkinsonismo atípico. Una de las quejas más frecuentes referidas por los pacientes son síntomas intestinales y urinarios. El estreñimiento puede llegar a ser intenso, complicándose en forma de megacolon o de seudoobstrucción intestinal11. Se debe a una reducción de la motilidad colónica, a consecuencia directa de la enfermedad, ya que en el plexo mientérico se ha descrito la presencia de cuerpos de Lewy. Las alteraciones urinarias incluyen urgencia miccional, nicturia y polaquiuria y, en las fases más avanzadas, incontinencia.
Las alteraciones en la función sexual, con disminución de la libido e impotencia en los varones, es una queja frecuente. Los pacientes pueden presentar hipotensión ortostática pero, si existe, suele cursar de forma asintomática. Las manifestaciones disautonómicas aparecen normalmente tras muchos años de evolución de la enfermedad.
Dolor y síntomas sensitivos
Los pacientes con EP se quejan de dolor y molestias sensitivas. Aproximadamente la mitad de los pacientes presentan dolor y hasta un 40% calambres en las piernas, entumecimiento y hormigueos, en algunos casos pueden preceder el inicio de las manifestaciones motoras20. Muchas veces estos síntomas pueden mejorar con el ajuste de la medicación antiparkinsoniana.
Manifestaciones infrecuentes de la enfermedad de Parkinson y su diagnóstico diferencial
El reconocimiento de la EP parece que no debería ser una tarea difícil. Sin embargo, sólo un 75% de los casos diagnosticados clínicamente, se confirman en la autopsia21. Esto se debe a que el síndrome de parkinsonismo puede ser producido por múltiples entidades (tabla 2). La historia clínica del paciente es decisiva, pues ayuda a identificar posibles causas de parkinsonismo secundario o sugerir un parkinsonismo atípico. Se debe preguntar con insistencia por los fármacos que toma el paciente, si hay historia de exposición a tóxicos y buscar antecedentes familiares de enfermedades neurológicas, incluyendo temblor esencial y EP. Se debe investigar la secuencia temporal precisa de la aparición de los síntomas, si éstos fueron de inicio unilateral o no. Si el paciente sigue tratamiento con l-DOPA, es importante saber si los síntomas respodieron a la levoterapia y en qué grado lo hicieron. Si existen síntomas o signos diferentes a los usuales o el curso y la progresión de la enfermedad es atípica habrá que descartar otras causas de parkinsonismo.

Inicio juvenil
Cuando la EP se inicia antes de los 40 años se define como «EP de inicio temprano», y se presenta con las mismas características que en los pacientes de más edad. La enfermedad de «inicio juvenil» (antes de los 20 años) se diferencia por una mayor frecuencia de historia familiar de EP y por la presencia de distonía al inicio22.
Para diagnosticar EP en menores de 40 años deben descartarse otras causas secundarias de parkinsonismo. Entre ellas destaca la enfermedad de Wilson, que es tratable y se diagnostica mediante determinaciones en plasma y orina derivadas del metabolismo del cobre y, mediante examen oftalmoscópico con lámpara de hendidura, la presencia de anillo de Kayser-Fleischer. Si existe enfermedad neurológica familiar deben descartarse la enfermedad de Huntington o la enfermedad de Machado-Joseph, que pueden cursar con parkinsonimo juvenil.
Temblor atípico o ausencia de temblor
La ausencia de temblor de reposo por historia o examen físico hace el diagnóstico de EP menos probable, aunque no lo descartan. Cuando esto sucede hay que pensar en otras causas de parkinsonismo y buscar signos característicos de estas entidades, como parálisis de la mirada vertical o distonía facial (PSP), hipotensión ortostática o ataxia de tronco (MSA), o signos piramidales (parkinsonismo de origen vascular). Sin embargo, los parkinsonismos atípicos también pueden presentar temblor de reposo. En la MSA puede observarse temblor de reposo en un 34% de los casos23 y en una serie de casos de PSP estaba presente en un 17%24.
El temblor esencial (TE) a menudo es incorrectamente orientado como EP. El TE es predominantemente postural y cinético, no se acompaña de rigidez o bradicinesia, y en un 50% de los casos es de carácter familiar. Hay que recordar que el fenómeno de «rueda dentada» puede observarse en el TE sin EP.
Un temblor irregular, claramente asimétrico y con mioclonías sugiere el diagnóstico de degeneración corti cobasal (DCB). Esta enfermedad se caracteriza por una afección motora progresiva asimétrica con parkinsonismo y signos motores y sensitivos que indican una
afección cortical focal. Otros hallazgos típicos incluyen distonía, mioclonías, apraxia y fenómeno de la mano ajena.
Inestabilidad postural
Aunque la inestabilidad postural es uno de los signos cardinales de la EP, la afección grave del equilibrio raramente se observa en los estadios iniciales de la enfermedad. En los casos donde predomina inicialmente un trastorno grave de la marcha deberán descartarse otras causas de parkinsonismo.
La PSP es una de las causas más frecuentes de parkinsonismo atípico. Uno de los síntomas iniciales son las caídas frecuentes. En estadios iniciales de la PSP, también podemos observar la afección de la mirada vertical, sobre todo hacia abajo, ausencia de temblor y nula respuesta a la l-DOPA.
La MSA también se presenta frecuentemente con una clara alteración postural. La MSA es un síndrome clínico caracterizado por déficit extrapiramidales como distonía, sobre todo cervical y facial, piramidales con hiperreflexia y signo de Babinski, cerebelosos y autonómicos. Los signos disautonómicos se manifiestan en forma de hipotensión postural, alteraciones del ritmo cardíaco, e impotencia en varones.
El parkinsonismo con una clara inestabilidad postural también puede ser una manifestación de la hidrocefalia normotensiva, que se puede acompañar de incontinencia urinaria y demencia de tipo subcortical.
Es importante recordar que el diagnóstico diferencial de la inestabilidad precoz debe incluir la misma EP. La alteración postural en la EP como síntoma precoz tiene importancia en su mal pronóstico, en cuanto a la progresión de la misma y la escasa respuesta a la
l-DOPA25.
Demencia al inicio de la enfermedad
La presencia de demencia al inicio de la enfermedad va en contra del diagnóstico de EP. Entre las demencias asociadas a parkinsonismo se incluyen EA, EDCL o la ECJ. También los cambios cognitivos en estadios iniciales de la enfermedad son frecuentes en la PSP, y
los pacientes pueden ser diagnosticados erróneamente de EA.
Neuroimagen en la enfermedad de Parkinson
Las técnicas morfológicas y funcionales de neuroimagen han tenido un importante desarrollo en los últimos años, y se han desarrollado diversos estudios que intentan definir las características de la neuroimagen de los pacientes con EP26. Los pacientes con EP y otros parkinsonismos presentan una disminución de la pars compacta de la SN, en secuencias T2 de la RM. También se ha observado que la existencia de hipointensidades putaminales está presente en otros parkinsonismos, del tipo de la PSP o la MSA, y en esta última, de forma más especifica, se observan hiperintensidades en los márgenes externos del putamen. La RM craneal puede ayudar a distinguir la EP de otros parkinsonismos atípicos.
Los estudios con tomografía por emisión de positrones (PET) con fluorodopa F18 o la tomografía con emisión de fotones han demostrado una reducción de la captación estriatal significativa respecto a controles, sobre todo en el putamen posterior, siendo la reducción directamente proporcional a la gravedad de la afección motora. En los pacientes con PSP, MSA o DCB, existe una disminución de la captación de fluorodopa F18 en el putamen y el caudado. Los estudios con PET que utilizan C-raclopride para evaluar el estado de los receptores postsinápticos, demuestran que la concentración de receptores D2 en la EP es normal o está aumentada, lo que le diferencia de otros parkinsonismos, como la PSP, donde la afección es postsináptica. La PET puede ayudar al diagnóstico de EP sintomática, y en ocasiones presintomática, y en el diagnóstico diferencial con otros parkinsonismos. El mayor inconveniente es el elevado coste económico y, a veces, su falta de disponibilidad.
Diagnóstico de la enfermedad de Parkinson
Aunque las técnicas de neuroimagen podrán ser en el futuro una herramienta útil en el diagnóstico de la EP, actualmente éste sigue realizándose mediante la anamnesis clínica, la exploración neurológica y el seguimiento del paciente. Los criterios más utilizados actualmente, que están basados en la combinación de estas manifestaciones clínicas, los propuestos por la UK Parkinson's Disease Society Brain Bank27 (tabla 3) o los más recientes de Gelb et al28 que mejoran la precisión diagnóstica. La confusión en el diagnóstico de la EP es mayor en estadios iniciales de la enfermedad, cuando muchos de los signos distintivos de la EP están ausentes.

Epidemiología
La EP es un trastorno neurodegenerativo progresivo que afecta a más del 1% de la población por encima de los 55 años. Su prevalencia aumenta con la edad, siendo del 3,1% en sujetos entre 75 y 84 años29. Constituye, pues, el trastorno neurodegenerativo más frecuente después de la enfermedad de Alzheimer. En Europa y Norteamérica se da la prevalencia más alta y en Asia y África la más baja30. La incidencia anual oscila entre 4,5 y 21 casos por cada 100.000 habitantes. La edad de inicio está entre los 55 y 60 años y, aproximadamente, dos tercios de los pacientes inician los síntomas entre los 50 y 70 años.
Etiología
Actualmente la causa de la EP es desconocida, aunque se están realizando importantes avances en las últimas décadas sobre la influencia de factores ambientales y genéticos en la EP.
Factores ambientales
Diversos estudios han encontrado una asociación entre la EP y determinados factores ambientales, como son: habitar en ambiente rural, consumo deagua de pozo, exposición a pesticidas y a preservativos de la madera (tabla 4). El consumo de tabaco se ha asociado de forma inversa al riesgo de sufrir la EP31. La exposición prolongada a cobre y manganeso o la exposición combinada a plomo, cobre y hierro, se han relacionado con mayor riesgo para presentar EP.

El parkinsonismo secundario a la intoxicación con 1-metil-4-fenil-1,2,3,6-tetrahidropiridina(MPTP) reforzó la hipótesis de la etiología tóxico-ambiental en la EP. Los primeros casos de parkinsonismo en intoxicados por MPTP fueron localizados en adictos a narcóticos por vía parenteral32. Los pacientes presentaban un cuadro clínico similar al descrito en la EP, con una instauración rápida de la sintomatología. El MPTP entra fácilmente en el SNC y es metabolizado en las células gliales, a través de la monoamino-oxidasa B (MAO B), en 1-metil-4-fenil-piridinio (MPP+), que es la toxina activa, siendo posteriormente recaptado por las neuronas nigroestriatales. El MPP+ se concentra en la mitocondria, inhibe al complejo I, disiminuye en la síntesis de ATP y produce un fallo energético celular que conduce a la muerte neuronal.
Factores genéticos
Además de la edad avanzada, la existencia de antecedentes familiares de EP o de temblor esencial constituyen los factores de riesgo principal para la EP33. En los últimos años, el estudio de mutaciones o polimorfismo en determinados genes hace que los factores genéticos influyan en la etiología de la EP.
Agregación familiar en la enfermedad de Parkinson
Se ha identificado la presencia de EP familiar entre el 13 y el 33% de los pacientes con EP33.
En un estudio con 18F-DOPA realizado en gemelos, uno de los cuales estaba afectado de EP, se reportó una disminución de la captación del marcador en los ganglios basales (compatible con EP presintomática) en el 45% de los gemelos «sanos» monozigotos frente al 29% de los dizigotos34. Otros estudios con PET han demostrado en varias familias con EP la existencia de afección subclínica en miembros no afectados.
Polimorfismos genéticos asociados a enfermedad de Parkinson
Los estudios de genética molecular se han orientado hacia la identificación de genes candidatos que pudieran asociarse a un riesgo aumentado de sufrir la EP. Se han investigado varios polimorfismos en genes que codifican para diversas enzimas relacionadas con el metabolismo de la dopamina y de otras sustancias.
La debrisoquina hidroxilasa es una enzima detoxificante. Por ello, defectos de esta enzima hacen que determinadas sustancias tóxicas no sean eliminadas y puedan producir daño neuronal en la SN.
La tirosina hidroxilasa (TH) cataliza el primer paso en la síntesis de catecolaminas o la conversión de L-tirosina en l-DOPA. Se han descrito varios casos de parkinsonismo de inicio en la infancia, con respuesta a
l-DOPA, asociados a mutaciones en el gen de la TH.
La MAO induce la formación de radicales libres tóxicos para la neurona. En concreto, la MAO-B transforma el MPTP en MPP+. Los estudios de asociación casos-controles en relación con polimorfismos de estas enzimas no han sido concluyentes35-41.
Una de las enzimas antioxidantes más importantes en el SNC es la cobre-cinc superóxido dismutasa (SOD1). Se ha reportado que la actividad SOD es mayor en pacientes con EP en estadios iniciales que en pacientes con EP en estadios avanzados. Esto supone una disminución de la actividad de la SOD con la evolución de la enfermedad. Sin embargo, en dos estudios diferentes no se han encontrado mutaciones en la SOD1 en casos familiares y esporádicos de EP42.
La alfa-1-antiquimotripsina (ACT) es una glucoproteína sérica que se une con gran afinidad al betaamiloide en el cerebro de los pacientes con EA. Recientemente, se ha demostrado una mayor frecuencia del genotipo ACT-AA en pacientes con EP de origen japonés comparados con controles sanos43. Sin embargo, en nuestra población se ha encontrado un porcentaje de portadores del genotipo AA similar en pacientes y controles, por lo que no parece que la ACT esté implicada en la etiología de la EP en nuestra población44.
Se ha reportado una frecuencia significativamente elevada de pacientes con EP homozigotos para el alelo 3 del receptor D2 de la dopamina (DRD2): 2,3 veces más frecuentes en los pacientes que en los controles. Esto sugería al principio una asociación entre dicho alelo y el desarrollo de la EP45. Sin embargo, en un estudio reciente en la población española, no se ha encontrado una diferencia significativa en la distribución alélica de un polimorfismo intrónico del gen del DRD2 entre pacientes y controles46. Tampoco los estudios realizados en la población japonesa han demostrado diferencias alélicas en la frecuencia de diferentes polimorfismos localizados en genes de los receptores D2, D3 y D4 en pacientes y controles sanos47. Por tanto, el papel de los genes de los receptores comparados con dopaminérgicos en la génesis de la EP está aún por definir. Recientemente, se ha descrito una asociación significativa del genotipo A0/A0 de un polimorfismo intrónico del gen de la proteína tau con la EP48.
Mutaciones causantes de enfermedad de Parkinson familiar
En los últimos años se han descrito en familias con EP diferentes mutaciones en dos genes se han asociado a EP familiar (tabla 5).

*-sinucleína (PARK1)
La *-sinucleína es una proteína que tiene un papel en la plasticidad neuronal. En humanos, la *-sinucleína ha sido implicada en la EA, la EP y la EDCL. En la EA, la *-sinucleína es un componente no betaamiloide de las placas seniles, por lo que se ha implicado en la formación de la placa amiloide49. En la EP y en la EDCL, la *-sinucleína se considera un componente fundamental de los cuerpos de Lewy, ya que éstos se tiñen con anticuerpos para *-sinucleína50. Además, recientemente, la *-sinucleína ha sido identificada en las inclusio nes cítoplasmáticas gliales y neuronales de pacientes con MSA, sugiriendo que el depósito de *-sinucleína está implicado en varias enfermedades neurodegenerativas.
Uno de los descubrimientos principales ha sido la detección de una mutación heterozigota en el gen de la *-sinucleína consistente en la sustitución de una treonina por una alanina en el residuo 53 (Ala53Thr) del exón 4 de la *-sinucleína en una familia de origen italiano, con múltiples miembros afectados a lo largo de varias generaciones, y en otras 3 familias griegas no relacionadas51. Este descubrimiento ha señalado que la *-sinucleína es el primer gen implicado directamente en la EP familiar. Un año más tarde, se detectó una nueva mutación en este gen (Ala30Pro) en una fami lia alemana52. Sin embargo, estas mutaciones sólo explican un número reducido de casos de EP con heren-
cia autosómica dominante, no habiéndose encontrado mutaciones el gen de la *-sinucleína en numerosos casos de EP familiar y esporádico en distintas poblaciones53,54.
Gen Parkin (PARK2)
El parkinsonismo juvenil autosómico recesivo (AR-JP) se caracteriza por una buena respuesta a la l-DOPA y un inicio en la edad juvenil, siendo la edad de media de inicio de 23 años. El gen responsable del AR-JP, Parkin, está situado en el locus cromosómico 6q25.2-27. Diversas mutaciones patogénicas en el gen Parkin como deleciones, sustituciones o multiplicaciones en varios exones de dicho gen, se han identificado en pacientes con EP familiar con herencia autosómica re cesiva en familias de origen japonés55, europeas, y africanas56. Esto sugiere que el espectro clínico de las mutaciones del gen Parkin es más amplio de lo que inicialmente se había supuesto. La enfermedad asociada a estas mutaciones se caracteriza por un curso relativamente benigno y la frecuente presentación inicial con distonía. Curiosamente, en el examen neuropatológico de algunos casos de AR-JP asociados a mutaciones del gen Parkin los cuerpos de Lewy están ausentes.
Otros locus cromosómicos asociados a la enfermedad de Parkinson familiar
Recientemente se han descrito dos nuevos locus para formas familares de EP: el primero (PARK3) localizado en el brazo corto del cromosoma 2 (2p13)57, y el segundo, en el brazo corto del cromosoma 4 (PARK4)58. Hasta la fecha, no han sido identificados los genes implicados en estas familias.
Sin embargo, estas mutaciones son responsables de la EP en un número reducido de familias. Todo ello hace pensar que nuevos genes todavía no localizados pueden causar EP familiar.
Nuevos fármacos en el tratamiento de la enfermedad de Parkinson
El tratamiento con l-DOPA mejora los síntomas clínicos y la calidad de vida de los pacientes parkinsonianos de forma notable. La l-DOPA sigue siendo el fármaco más potente y efectivo para el control de los síntomas de la EP, pero estos beneficios están muy limitados por los efectos secundarios que suceden a medio y largo plazo tras pocos años del inicio del tratamiento. La l-DOPA administrada crónicamente ocasiona fluctuaciones motoras y discinesias en muchos pacientes. Las fluctuaciones motoras son períodos, de duración variable, en los que reaparecen los síntomas parkinsonianos (períodos off). Las discinesias consisten en movimientos anormales involuntarios de tipo generalmente coreico o distónico que aparecen cuando la l-DOPA inicia o finaliza su acción (discinesias de comienzo o final de dosis) o durante el período on (discinesias de beneficio). En la fisiopatología de estas complicaciones motoras se han implicado los cambios degenerativos de las neuronas dopaminérgicas de la SN, que pierden la capacidad de almacenamiento de dopamina. Se han implicado en el origen de las discinesia cambios bioquímicos en los receptores dopaminérgicos postsinápticos estriatales59.
Para la prevención y el tratamiento de las complicaciones motoras de la levoterapia, disponemos de nuevos fármacos efectivos.
Inhibidores de la catecol-O-metil transferasa
La catecol-O-metil transferasa (COMT) es una enzima que interviene en el metabolismo de catecolaminas como la dopamina, noradrenalina, adrenalina y sus metabolitos, como l-DOPA. Cuando la carbidopa o la benseracida inhiben la decarboxilación de la l-DOPA a dopamina, la COMT convierte la mayor parte de la l-DOPA en la periferia en 3-O-metil-l-DOPA (3-OMD). La inhibición periférica de la COMT permite incrementar la cantidad de l-DOPA disponible en el cerebro y, por tanto, incrementar la neurotransmisión dopaminérgica a nivel cerebral. Los inhibidores de la COMT (ICOMT) actúan principalmente en el aumento de la vida media de la l-DOPA. Por ello, en los pacientes con fluctuaciones motoras, la inhibición de la COMT prolonga la respuesta de la l-DOPA sin aumentar excesivamente las discinesias u otros efectos secundarios60.
Los ICOMT más conocidos son el tolcapone y el entacapone. El tolcapone ha demostrado en los estudios clínicos una notable eficacia terapéutica; sin embargo, la aparición de algunos casos de hepatitis fulminan te en pacientes que tomaban tolcapone, forzó la retirada de este medicamento en Europa. El entacapone dispone sólo de actividad periférica. También ha demostrado en estudios controlados un aumento de los períodos de efectividad de la l-DOPA (períodos on) y permite una reducción de l-DOPA en algunos pacientes, aunque también produce un aumento en la duración de las discinesias61. Los ICOMT son fármacos, en general, bien tolerados y entre sus efectos colaterales destacan la diarrea (1-3% de los casos) y las alucinaciones.
Nuevos agonistas dopaminérgicos
Tras la eficacia terapéutica demostrada durante años por la bromocriptina, han surgido posteriormente diversos agonistas de los receptores dopaminérgicos, como pergolida, lisurida y apomorfina, entre otros. El de sarrollo farmacológico actual ha permitido la aparición de nuevos agonistas de los receptores dopaminérgicos con ventajas farmacocinéticas. En la tabla 6 se detallan las características de los distintos receptores. Aunque los agonistas D2 son los que han demostrado ser más eficaces hasta la actualidad, los agonistas D1 puros, aunque aún no comercializados, se están investigando activamente. Sin embargo, queremos destacar tres agonistas dopaminérgicos de reciente aparición: el pramipexole, el ropinirole y la cabergolina.

El pramipexole es un agonista dopaminérgico no ergótico, activo por vía oral, que posee una actividad selectiva sobre los receptores dopaminérgicos de la subfamilia D2 (D2, D3 y D4) y con afinidad preferente por los receptores D3. Está aprobado como monoterapia para el tratamiento inicial de la EP y como tratamiento coadyuvante a la l-DOPA en pacientes con enfermedad avanzada62. Los datos obtenidos de estudios de duración relativamente larga sugieren que la monoterapia con pramipexole mejora las actividades de la vida diaria y los síntomas motores en la EP inicial. Como el resto de los otros agonistas, los efectos adversos más frecuentes que produce son náuseas, mareos, somnolencia, insomnio, estreñimiento, astenia y alucinaciones.
El ropinirole posee también una afinidad exclusiva por los receptores D2 con estructura no ergólica que, como el pramipexole, es eficaz en monoterapia de la EP inicial. Se ha evidenciado una capacidad similar a la observada con l-DOPA para revertir los síntomas parkinsonianos en estadios iniciales de la enfermedad, aunque en estadios avanzados la eficacia de la l-DOPA es superior a la del ropinirole63. Asimismo, su administración como coadyuvante mejora las fluctuaciones motoras y permite una reducción de las dosis de l-DOPA.
La cabergolina es un potente agonista D2 con una vida media superior a 65 h, por lo que una única dosis al día produce una estimulación dopaminérgica continua. Se ha demostrado su eficacia en la EP inicial, durante un período de tiempo superior a un año, siendo sólo marginalmente menos efectiva que la l-DOPA64.
Finalmente, el único agonista de acción rápida, la apomorfina, que actúa sobre los receptores D1 y D2, se puede administrar por vía subcutanea, intranasal, sublingual y rectal. Numerosos estudios han verificado su eficacia en el tratamiento de las fluctuaciones motoras por vía subcutánea y transnasal65. A su vez, estudios recientes han revelado la posibilidad de administrarlo vía transdérmica66. Los pacientes con fluctuaciones motoras de larga duración, incapacitantes y resistentes a múltiples estrategias farmacológicas, se pueden beneficiar de la apomorfina.
Antagonistas del glutamato
Recientemente, se han estudiado los efectos de la amantadina, y también los efectos del antagonista no competitivo de los receptores NMDA dextrometorfano, sobre las fluctuaciones motoras y discinesias en pacientes afectados de EP avanzada. Estudios a doble ciego realizados con ambos fármacos presentaron un efecto antidiscinético de ambos y, además, la amantadina mejoró las fluctuaciones motoras67,68. Los resultados de estos estudios apoyan la hipótesis de que la hiperactivación glutaminérgica es responsable de algunas complicaciones motoras asociadas al tratamiento con l-DOPA. Sin embargo, estudios con otros fármacos antiglutamatérgicos, como lamotrigina, no han presentado efectos antiparkinsonianos claros.
Nuevos inhibidores de la monoamino oxidasa (MAO)
La MAO, con sus dos isoformas MAO-A y MAO-B, es la principal enzima cerebral relacionada con el catabolismo intracelular de monoaminas. La selegilina, un inhibidor de la MAO-B, se ha utilizado en los últimos años en el tratamiento de la EP con el objetivo de lograr un efecto neuroprotector, aunque este hecho no se ha podido demostrar69. Este fármaco, no obstante, presenta un cierto efecto sintomático antiparkinsoniano.
Recientemente, se han desarrollados nuevos fármacos de esta familia. La lazabemida es un inhibidor reversible de la MAO-B, 100 veces más selectivo que la selegilina en el bloqueo enzimático. A diferencia de la se legilina, la lazabemida no se metaboliza en compuestos potencialmente peligrosos como la L-anfetamina. Se ha demostrado un teórico poder preventivo de la lazabemida en la producción de radicales libres en el cerebro70. Sin embargo, los estudios clínicos realizados con lazabemida han dado lugar a resultados contradictorios.
La moclobemida es un antidepresivo inhibidor rever sible de la MAO-A. En un estudio no controlado en 12 pacientes con EP sin síntomas depresivos, se observó una mayor rapidez y duración de la acción de la l-DOPA. Se observó, sin embargo, hipotensión importante, que fue sintomática en uno de los pacientes. Más recientemente, en un estudio abierto con 20 pacientes no deprimidos con EP que presentaban complicaciones motoras, la administración de moclobemida se redujo en un 27% el tiempo en off, sin modificar el período on71. Otros estudios encontraron un empeoramiento de las discinesias72.
Antipsicóticos atípicos
En el tratamiento de los síntomas psiquiátricos, como alucinaciones o psicosis, que pueden aparecer como complicación del tratamiento crónico con l-DOPA se utilizan los neurolépticos atípicos. Éstos mejoran los síntomas psiquiátricos sin que se produzca un deterioro de los síntomas extrapiramidales en la EP, a diferencia de los neurolépticos clásicos. Esto es debido a que los neurolépticos atípicos tienen propiedades farmacocinéticas especiales, como la ausencia de hiperregulación de los receptores dopaminérgicos o el hecho de que no incrementan la secreción de prolactina. La clozapina es el más utilizado dada su gran eficacia. Posee una gran afinidad por los receptores D1, D3 y D4 y una baja afinidad por los receptores D2, por lo que el bloqueo dopaminérgico se produce sólo a nivel del sistema límbico. Diversos estudios han demostrado la eficacia de la clozapina a dosis bajas en el tratamiento de la psicosis dopaminérgica73. El efecto secundario más frecuente es la sedación. La granulocitopenia puede observarse en un 1,5-3% de los pacientes durante el tratamiento, pero la agranulocitosis se presenta en el 0,8% de pacientes/ año, por lo que la monitorización continuada del hemograma es necesaria.
Otros neurolépticos atípicos recientes son la risperidona y la olanzapina. La risperidona es un derivado benzisoxazólico con un potente antagonismo por los receptores serotoninérgicos 5-HT2 y dopaminérgicos D2, y en menor cuantía por los D4 y D3. Los beneficios clínicos producidos por este fármaco en pacientes con EP y psicosis dopaminérgica no están claramente demostrados.
La olanzapina es un derivado con afinidad por los receptores 5-HT 2A/2C, dopaminérgicos D1, D2 y D4, muscarínicos, *-1 adrenérgicos e histamínicos H1. No se han observado alteraciones hematológicas, aunque la eficacia de la olanzapina es menor que la clozapina74.
Recientemente se han publicado estudios clínicos con un nuevo antipsicótico atípico, la quetiapina, que ha ofrecido buenos resultados pero inferiores a los de la clozapina75.
Tratamiento quirúrgico de la enfermedad de Parkinson
El tratamiento quirúrgico de la EP se inició en los años 4076. El advenimiento de la l-DOPA desplazó los procedimientos neuroquirúrgicos utilizados hasta que han recobrado interés en la última década. Actualmente, se está empleando la neurocirugía funcional en la EP para el tratamiento de las fluctuaciones motoras y las discinesias, sin aparente solución farmacológica.
En la actualidad, existen dos tipos de tratamiento quirúrgico para la EP: la cirugía funcional y las técnicas de «restauración» neuronal. La neurocirugía funcional consiste en la ablación o la estimulación de núcleos cerebrales. Las técnicas de «restauración» neuronal intentan corregir el defecto bioquímico inicial de la EP, la pérdida de dopamina, añadiendo células dopaminérgicas o promoviendo la supervivencia de las células remanentes. En este grupo se incluyen procedimientos como el trasplante de células dopaminérgicas y la infusión intratecal de factores de crecimiento o neurotró-
ficos.
Cirugía funcional en la enfermedad de Parkinson
Funcionamiento de los ganglios basales y bases anatómicas de la cirugía funcional. Los ganglios de la base son un conjunto de núcleos cerebrales cuyo funcionamiento se halla modulado por la dopamina procedente de la SN77. Los ganglios basales están compuestos por los siguientes núcleos: putamen, caudado, globo pálido (interno y externo; GPi, GPe), subtalámo (NST) y SN. En la EP, el déficit de dopamina condiciona un funcionamiento inadecuado de estas estructuras, que se traduce en una activación excesiva de dos de sus núcleos: el NST y el GPi78. La hiperactividad de estos núcleos condiciona un aumento de la inhibición del tálamo, que inhibe la corteza premotora, responsable de los síntomas de la EP.
La cirugía funcional de la EP trata de normalizar esta actividad alterada de estos núcleos a través de la lesión de los núcleos que están hiperactivados (GPi, NST). Esto se realiza mediante la palidotomía y la subtalamotomía, o mediante la neuroinhibición de estos núcleos empleando una estimulación directa (palidal y subtalámica). La lesión de estos núcleos se realiza a través de la termocoagulación y se suele utilizar un microrregistro neuronal para determinar mejor la localización de los núcleos cerebrales. La neuroinhibición se realiza a través de la implantación de un electrodo, que produce estímulos eléctricos a elevada frecuencia79.
Pacientes candidatos de tratamiento quirúrgico. Los pacientes candidatos a ser intervenidos son aquellos con criterios de EP idiopática, con edades entre 35 y 70 años, con importante incapacidad funcional y con presencia de fluctuaciones motoras y discinesias, que no responde al tratamiento farmacológico adecuado para el estadio de la enfermedad.No deben operarse los pacientes con enfermedades generales o neurológicas que contraindiquen la cirugía, los pacientes con deterioro cognitivo o alteraciones psicoafectivas que impidan una adecuada colaboración durante la intervención o, por motivos técnicos, los pacientes con clara atrofia cerebral en los estudios de neuroimagen.
Dianas quirúrgicas empleadas actualmente en cirugía funcional. La diana quirúrgica en la cirugía funcional se localiza en los núcleos cerebrales profundos que presentan un funcionamiento anormal en la EP: el tálamo, el GPi y el NST. Sobre estos núcleos se emplean tanto técnicas ablativas como de estimulación. No obstante, sobre el NST, dado el riesgo de hemibalismo poslesional, se aplican fundamentalmente técnicas de estimulación, hallándose la subtalamotomía en una fase experimental80.
Talamotomía y estimulación talámica. La talamotomía consiste en la termocoagulación del núcleo ventral intermedio del tálamo (VIM) y produce una mejoría del temblor parkinsoniano. La mejoría es del 86-100% y
se mantiene tras 10 años del procedimiento81. La talamotomía bilateral mejora el temblor bilateral pero produce disartria e hipofonía graves en un gran número de
casos.
La estimulación talámica, que se realiza sobre todo a nivel del VIM, mejora el temblor. La tasa de respuesta es del 88%, que se mantiene tras 8 años tras la intervención82. El resto de síntomas parkinsonianos no mejoran con la estimulación talámica. El síntoma secundario a la intervención más frecuente es la disartria.
Palidotomía y estimulación palidal. La palidotomía se ha realizado a través de la termocoagulación del GPi y en la mayoría se han utilizado técnicas de microrregistro de la actividad neuronal. El porcentaje de cambio observado un año tras la cirugía en la puntuación total de la escala unificada para la EP fue entre 16 y el 25%, y en la subescala motora entre el 30 y el 60%. A su vez, dos estudios realizados en nuestro medio indicaron, un añodespués de la intervención, una mejoría de los síntomas motores de la enfermedad, valorados en estado «off», en torno al 30%83. Esta mejoría, estadísticamente significativa, es consecuencia de la recuperación observada en los síntomas cardinales de la enfermedad en las extremidades contralaterales al GPi intervenido; el temblor disminuyó un 48%, la rigidez un 36,2% y la bradicinesia un 37,4%. Resultados similares han sido publicados por otros grupos. Un efecto importante de la palidotomía es la eliminación de las discinesias contralaterales al hemisferio intervenido. Todos estos cambios motores son capaces de mejorar de forma significativa la calidad de vida de estos pacientes 3 meses tras la intervención84. Sin embargo, la dosis de medicación antiparkinsoniana no se modificó significativamente tras la intervención. Las complicaciones más frecuentes de la palidotomía son la hemianopsia homónima, la paresia facial y la hemiparesia, siendo la frecuencia de aparición de los mismos muy variable.
La palidotomia bilateral no ha sido convenientemente estudiada, debido a los efectos adversos graves de las intervenciones realizadas, por lo que no está recomendada actualmente.
La estimulación palidal ofrece resultados similares a los descritos con la palidotomía. Al tratarse de un procedimiento no ablativo puede aplicarse bilateralmente, proporcionando una mayor reducción de los síntomas axiales en el período off. En estado on, al igual que la palidotomía, mejora las discinesias, aunque se han descrito empeoramientos de algunos síntomas parkinsonianos durante las fases on85. Los efectos adversos descritos son la aparición de fenómenos positivos visuales (fosfenos) y las alteraciones del lenguaje.
Estimulación bilateral del núcleo subtalámico. La estimulación bilateral del NST mejora todos los síntomas cardinales de la EP y las actividades cotidianas al menos un 60%, en un seguimiento de un año; en más del 50% se reduce la medicación y disminuyen las discinesias86. Los efectos colaterales poco frecuentes son: empeoramiento cognitivo transitorio, disartria e hipofonía. Siempre existe el riesgo de todo procedimiento neuroquirúrgico, como son los de hematomas cerebrales, que si bien son infrecuentes pueden llegar a ser mortales.
En nuestro medio los resultados obtenidos tras la estimulación bilateral del NST en 15 pacientes a los 6 meses de la intervención1 incluyen una mejoría de los síntomas parkinsonianos en torno al 70%, con una reducción de la medicación superior al 75%. Nueve de estos 15 pacientes pudieron dejar de tomar su medicación antiparkinsoniana sin empeoramiento en las activi dades cotidianas. Los efectos secundarios incluyeron desorientación transitoria en 2 pacientes y disartria e hipofonía persistentes al año de seguimiento en otros dos pacientes. En la figura 1 aparece la colocación de 2 estimuladores en ambos núcleos subtalámicos.
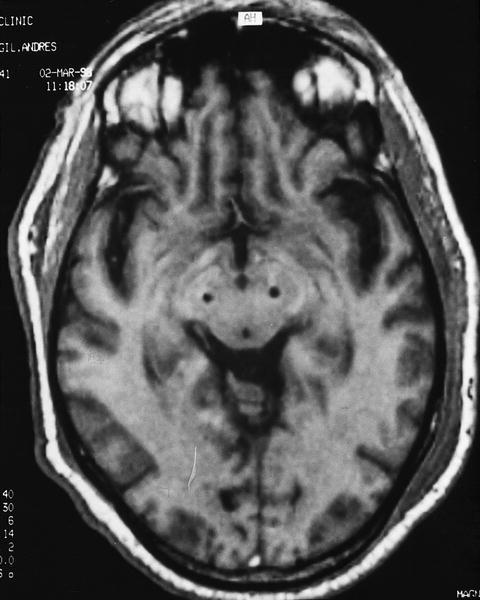
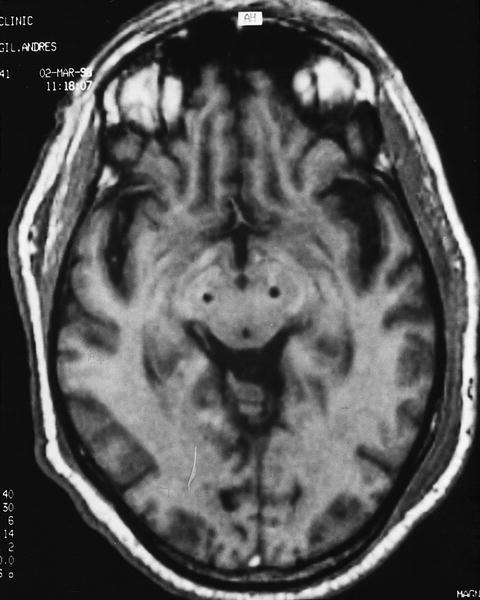
Fig. 1. Corte axial de una RM craneal que presenta la situación de los electrodos en ambos núcleos subtalámicos para estimulación bilateral en un paciente con enfermedad de Parkinson (cortesía del Dr. Valldeoriola).
Dada la moderada mejoría obtenida sobre la bradicinesia y la rigidez, la cirugía sobre el tálamo (ablación y estimulación) está siendo desplazada por la cirugía hacia otras dianas cerebrales. De manera general, la estimulación ofrece ventajas sobre los procesos ablativos. En cuanto a la estimulación bilateral, la diana de elección (GPi o NST) no está plenamente decidida. Como ya se ha expuesto previamente, la estimulación bilateral del NST parece mejorar todos los síntomas cardinales de la EP y permite reducir de forma importante el tratamiento85.
«Restauración» neuronal en la enfermedad de Parkinson
Trasplante de células. La pérdida de neuronas productoras de dopamina en la SNpc es el defecto original de la disfunción del resto de núcleos de los ganglios basales en la EP. El objetivo del trasplante neuronal es reemplazar las células dopaminérgicas de la SN implantándolas directamente en el cerebro. Hasta la fecha se han realizado en enfermos dos procedimientos de implantación de células que producen dopamina.
Los trasplantes autólogos de células cromafines de la médula suprarrenal productoras de dopamina y de otros factores tróficos neuronales en núcleo caudado han sido capaces de producir en la mayoría de las series una mejoría moderada o pasajera, aunque sólo una serie refiere una mejoría importante de los pacientes87,88.
Los trasplantes de neuronas dopaminérgicas de SN fetal han demostrado, en general, una mejoría discreta de los síntomas con resultados muy heterogéneos. Además, el procedimiento tiene inconvenientes éticos importantes para muchos investigadores89,90.
Otras técnicas de trasplante en experimentación serían la transformación genética de fibroblastos y de células musculares para que produzcan dopamina. También se está realizando el autotrasplante de células noradrenérgicas del cuerpo carotídeo en monos. Dichos injertos sobreviven tras ser implantados en estriado y mejoran los síntomas parkinsonianos91. La inyección intratecal de factores neurotróficos derivados de las células gliales se está experimentando actualmente.