




La hemorragia digestiva es un problema muy común dentro de la práctica clínica habitual y una causa importante de morbimortalidad. Se estima que su incidencia en nuestro medio es superior a los 150 casos por cada 100.000 habitantes y año, mientras las cifras de mortalidad rondan el 10% en las diferentes series. Este notable porcentaje se ha mantenido relativamente constante a lo largo de las últimas décadas, a pesar de los avances diagnósticos y terapéuticos, lo que se explica por la aparición de esta enfermedad en poblaciones progresivamente más envejecidas, con procesos comórbidos añadidos y por el creciente consumo de fármacos gastrolesivos1.
En función de su localización inicial, la hemorragia digestiva ha sido clásicamente dividida en dos entidades: la hemorragia digestiva alta y la hemorragia digestiva baja, según la lesión responsable se sitúe por encima o por debajo del ángulo de Treitz o duodenoyeyunal. Se trata de una clasificación no sólo con interés topográfico, sino también con implicaciones en la clínica, el tratamiento y el pronóstico. Concebiendo el problema en su conjunto, puede decirse que, excluyendo el origen hemorroidal, la principal causa de hemorragia digestiva en países occidentales es la enfermedad péptica, seguida a distancia por otras como las lesiones secundarias a hipertensión portal, la enfermedad diverticular y las neoplasias2.
La hemorragia digestiva alta (HDA) es responsable de la hospitalización de 100 pacientes por cada 100.000 habitantes y año, y de una mortalidad que oscila entre el 5 y el 12% de los casos. Su origen más frecuente es, como se ha comentado, la enfermedad péptica, término que engloba a la úlcera gastroduodenal, las lesiones agudas de la mucosa gástrica y la enfermedad por reflujo gastroesofágico. A estas causas de HDA siguen otras, como las varices esofágicas (la que posee mayor índice de mortalidad), el síndrome de Mallory Weiss, las neoplasias y la hemobilia. Finalmente, existe un grupo etiológico infrecuente constituido por coagulopatías, vasculopatías y parasitosis, cuyo interés radica esencialmente en su relación con enfermedades sistémicas, en las que la hemorragia digestiva es una manifestación más dentro de un amplio abanico de signos y síntomas. Clínicamente, la HDA suele presentarse en forma de melenas y/o hematemesis, aunque cuando el tránsito intestinal es rápido puede hacerlo en forma de hematoquecia. Su diagnóstico se apoya básicamente en el empleo de la endoscopia digestiva alta. El tratamiento ha sido perfeccionado en las últimas décadas gracias a los avances técnicos endoscópicos y a la terapia erradicadora para Helicobacter pylori, lo que se ha traducido en una mejora en el pronóstico de estos pacientes2-5.
La hemorragia digestiva baja (HDB) es la causa del ingreso hospitalario en alrededor de 25 pacientes por cada 100.000 habitantes y año, y cuenta con una mortalidad inferior al 5% aunque, cuando la hemorragia es masiva, esta cifra puede verse incrementada hasta alcanzar el 21%. La etiología en los países occidentales varía en función de la edad del paciente. En niños y adolescentes, las causas más frecuentes son las fisuras anales, el divertículo de Meckel, la enfermedad inflamatoria intestinal y la poliposis juvenil. En ancianos, en los que esta forma de hemorragia digestiva es mucho más frecuente, deben ser consideradas de inicio la presencia de enfermedad hemorroidal, la diverticulosis, la angiodisplasia y el cáncer colorrectal. Las manifestaciones clínicas esenciales de la HDB son la rectorragia y la hematoquecia, aunque si la sangre procede del intestino delgado distal o del colon derecho puede adquirir forma de melena. En la mayoría de los casos, la hemorragia se autolimita y sólo en el 10-15% de éstos posee carácter persistente o recidivante. El diagnóstico definitivo suele requerir la realización de una fibrocolonoscopia, pero en algunos episodios de mínimo débito hemorrágico se precisa la utilización de métodos como la angiografía o la gammagrafía. Su tratamiento es muy variable y se define en función de la causa2-5.
Existen enfermedades que cursan con HDA y/o HDB que se acompañan de lesiones cutáneas y que pueden orientar el diagnóstico mediante una simple exploración física (tabla 1). Estas manifestaciones cutáneas pueden preceder a la hemorragia digestiva, lo que ofrecería la opción de realizar una profilaxis adecuada o un tratamiento precoz; también pueden aparecer simultáneamente, contribuyendo de una manera muy útil al diagnóstico etiológico. Por estas razones, la detección de estas dermatosis es una herramienta que el clínico debe conocer y manejar para conseguir un mayor rendimiento diagnóstico y terapéutico. En el presente artículo trataremos las dermatosis asociadas a la hemorragia digestiva más frecuentes, agrupándolas en función de su mecanismo fisiopatológico.

Anomalías congénitas de los vasos sanguíneos
Enfermedad de Rendu-Osler-Weber
La enfermedad de Rendu-Osler-Weber o telangiectasia hemorrágica hereditaria (THH) es una displasia fibrovascular sistémica que se hereda con carácter autosómico dominante, aunque en un 18-46% de casos no existe historia familiar conocida6. Su prevalencia es de 1-2 casos por 100.000 habitantes7 y se caracteriza por la presencia de telangiectasias cutáneas, mucosas y viscerales, pudiéndose presentar también malformaciones arteriovenosas y aneurismas, con una tendencia hemorrágica recidivante8.
La manifestación clínica más frecuente de la THH es la epistaxis, que aparece en el 80% de los casos y es a menudo la forma de presentación de la enfermedad. Suele iniciarse en la edad infantil. La hemorragia gastrointestinal es la segunda manifestación clínica en frecuencia (13-40% de casos) y suele darse en la quinta década de la vida7. Tanto las epistaxis repetidas como las pérdidas digestivas pueden ser responsables de anemias ferropénicas, en muchos casos graves, que pueden ser causa de muerte, aunque de forma poco frecuente7,9.
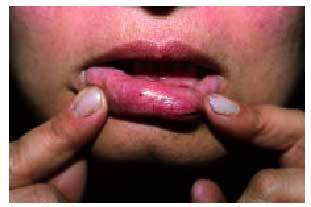
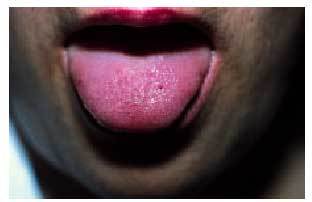
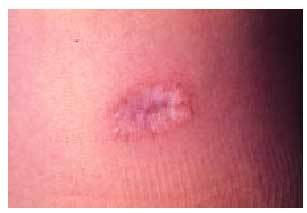
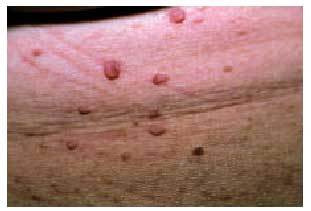
Desde el punto de vista dermatológico, el hallazgo clínico más frecuente son las telangiectasias, puntiformes o lineales, localizadas sobre todo en la mitad superior del cuerpo (cara, labios, lengua, paladar, nariz, orejas, manos y tórax) (figs. 1 y 2). Suelen aparecer en torno a la tercera o cuarta décadas de la vida. Desde el punto de vista histológico, encontramos capilares irregularmente dilatados en la dermis papilar, con cambios degenerativos en el tejido de soporte perivascular8.
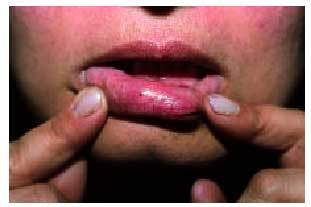
Fig. 1. Telangiectasias en la semimucosa labial en una paciente con enfermedad de Rendu-Osler-Weber.
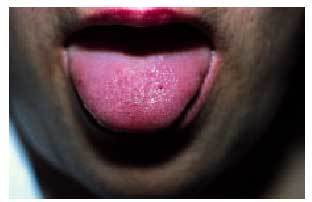
Fig. 2. Telangiectasias en la lengua en la paciente de la figura 1 (enfermedad de Rendu-Osler-Weber).
La THH es causa también de anastomosis arteriovenosas pulmonares y hepáticas10, aneurismas del arco aórtico y la arteria esplénica y, con menor frecuencia, lesiones oculares y del sistema nervioso central (SNC).
El tratamiento suele limitarse al control de la anemia secundaria a la hemorragia, aunque puede requerirse la electrocirugía o laserterpia endoscópica y la cirugía para casos de afección gastrointestinal intensa. Para las lesiones nasales que provoquen epistaxis intensas, pue den utilizarse estrógenos tópicos11, laserterapia, embolizaciones intraarteriales supraselectivas12 o injertos cutáneos. La electrocirugía y el láser tienen utilidad cosmética en el caso de las lesiones cutáneas.
El pronóstico, en general, es bueno, aunque existe una mortalidad inferior al 10%, relacionada directamente con la enfermedad.
Hemangiomatosis neonatal difusa
La hemangiomatosis neonatal difusa (HND) es un proceso raro, de herencia autosómica dominante, que asocia múltiples angiomas tuberosos de pequeño tamaño en el tegumento cutáneo, los órganos internos y el SNC. Las lesiones aparecen en el nacimiento o se desarrollan en las primeras semanas de la vida13,14. Los índices de morbimortalidad son elevados en esta entidad.
Los hemangiomas cutáneos son lesiones de 2 mm a 2 cm de diámetro mayor, de coloración rojo fresa a rojo oscuro, cuyo número puede oscilar entre unas pocas lesiones a cientos de ellas, pudiendo afectar la totalidad del tegumento.
El hígado es el órgano interno más frecuentemente afectado, seguido del pulmón, el cerebro y el intestino. Igualmente, se han descrito alteraciones en el estómago, el páncreas, los ovarios, la vagina y los ojos.
En ocasiones, se produce una involución de los hemangiomas cutáneos e incluso hepáticos. Cuando esto no ocurre, pueden presentarse serias complicaciones, sobre todo durante la fase de proliferación de las lesiones15. La mayoría de los pacientes con afección importante del hígado presentan hepatomegalia, fallo cardíaco y anemia. La causa más frecuente de muerte es el fallo cardíaco por fístulas arteriovenosas, que representa entre el 60 y el 90% de los casos en los primeros meses de vida. Otras complicaciones son las hemorragias gastrointestinales y cerebrales, que pueden ocasionar secuelas importantes16.
El diagnóstico de esta entidad se basa en la clínica cutánea, de manera que, en pacientes menores de 3 meses que presenten numerosos hemangiomas cutáneos es necesario realizar estudios complementarios radiológicos en busca de alteraciones viscerales17.
En general, el tratamiento debe de ser conservador. Se han utilizado tratamientos con corticoides sistémicos, interferón alfa-2, láser e incluso asociaciones de todos ellos, siendo los resultados inciertos. En ocasiones es necesaria la cirugía, siendo ésta de gran complejidad. Sin tratamiento, la muerte sobreviene en pocos meses18.
Defectos hereditarios del tejido conectivo
Seudoxantoma elástico
El seudoxantoma elástico (SE) es una enfermedad he-reditaria infrecuente que se caracteriza por una degeneración generalizada de las fibras elásticas, y cuya prevalencia es de 1/160.000 adultos19. La enfermedad fue descrita por Rigal en 1881 y Balzer en 1884, pero fueron Gronblaud y Strandberg en 192920 quienes relacionaron las lesiones cutáneas con las oculares, dando un carácter sistémico a este cuadro. Desde entonces, se establece de forma precisa sus características clínicas consistentes en manifestaciones cutáneas, alteraciones oculares y lesiones cardiovasculares.
La etiología del SE es desconocida y el defecto genético molecular subyacente está por determinar. La enfermedad presenta heterogeneidad genética, con la existencia de patrones autosómicos dominantes y recesivos21. Es más frecuente en mujeres y suele manifestarse a partir de la segunda década de la vida.
Dentro de las variadas manifestaciones que puede presentar el SE, las cutáneas son las más características de la enfermedad, las más frecuentes (presentes en el 80% de los casos) y las primeras que se suelen reconocer. Las lesiones cutáneas consisten en pápulas de coloración amarillenta o anaranjada, entre 1 y 5 mm de diámetro, redondeadas u ovales que se agrupan formando placas (aspecto «en piel de naranja»). La piel afectada presenta una superficie irregular, discretamente arrugada y de consistencia flácida y laxa. Las lesiones suelen distribuirse de forma bilateral y simétrica afectando preferentemente las áreas de flexuras (zona lateral de cuello, axilas, pliegue antecubital e ingles). Pueden apreciarse lesiones en las mucosas, fundamentalmente en el labio inferior, la mucosa oral y el paladar blando.
La hemorragia gastrointestinal en el SE ocurre aproximadamente en el 13% de los pacientes, afectando sobre todo a personas jóvenes (entre la segunda y la tercera décadas de la vida) y pudiendo ser la primera manifestación de la enfermedad22. Suele manifestarse como HDA, siendo la HDB menos frecuente. En la endoscopia de estos pacientes se aprecia una mucosa con pápulas amarillentas similares a las halladas en la piel, que pueden sangrar «en sábana»23.
Las manifestaciones oculares y arteriales son importantes en el SE. Las alteraciones oculares más frecuentes y/o características son las estrías angioides secundarias a la rotura de la membrana de Bruch, pigmentaciones moteadas de la retina o «manchas de leopardo», hemorragias retineanas y coroiditis, que pueden conducir a la ceguera24. La afección vascular se puede manifestar en forma de HTA, claudicación intermitente, abolición de pulsos periféricos, cardiopatía isquémica y hemorragia cerebral, incidiendo sobre todo en las arterias de mayor tamaño y las viscerales.
El diagnóstico se basa en la clínica y los hallazgos histopatológicos. En la biopsia cutánea, se observa una calcificación y fragmentación de las fibras elásticas en la zona media de la dermis reticular25.
La evolución del SE es gradualmente progresiva y el pronóstico está ligado a la afección visceral, especialmente del sistema cardiovascular. En el 70% de los pacientes con afección ocular se produce una limitación grave de la visión que puede conducir a la ceguera.
En la actualidad no existe tratamiento específico. Es importante realizar un consejo genético siempre que pueda establecerse el patrón de herencia. Pueden corregirse las lesiones cutáneas quirúrgicamente. Las lesiones de la retina requieren control periódico por parte del oftalmólogo y en caso de hemorragia hay que valorar la coagulación con láser. Muchas de las hemorragias gástricas remiten espontáneamente con tratamiento conservador, aunque en ocasiones es necesaria la cirugía, realizándose vagotomías con piloroplastia (útiles sobre todo en mujeres embarazadas) y gastrectomías totales26.
Síndrome de Ehlers-Danlos
Es una distrofia hereditaria compleja, caracterizada por hiperelasticidad de la piel, fragilidad cutánea y vascular e hiperlaxitud articular, causada por alteraciones de las fibras de colágeno. En la actualidad existen 10 clases, dependiendo del tipo de herencia, las alteraciones bioquímicas y las características clínicas.
De los diez tipos que existen, tan sólo el tipo IV (forma arterial o equimótica) presenta un considerable riesgo de hemorragia digestiva27. Es una forma grave, aunque poco frecuente, de síndrome de Ehlers-Danlos que se puede heredar de manera autosómica dominante o recesiva y se debe a un defecto en la síntesis de colágeno III causada, a su vez, por una mutación en el gen COL3A128.
Presenta escasas alteraciones cutáneas y articulares; sin embargo, tiene un riesgo aumentado de rotura arterial, intestinal (más frecuente en el colon sigmoide) y uterina, que pueden ser causa de mortalidad. La supervivencia media de estos pacientes se sitúa en torno a los 48 años29.
Los datos clínicos que nos pueden sugerir un síndrome de Ehlers-Danlos son: a) hiperelasticidad de la piel, signo más llamativo y constante, advirtiéndose sobre todo en los codos, la superficie posterior de los brazos, las rodillas, el cuello y las mejillas; b) hiperelasticidad articular, haciendo factibles actitudes acrobáticas de los «hombres de goma» que se exhibían en circos y ferias; c) fragilidad cutánea o dermorrexis, que provoca cicatrices atróficas en los codos, las rodillas y otras superficies fácilmente traumatizadas, así como placas de piel muy delgadas con aspecto papiráceo, y d) tumores seudomoluscoides blanco-azulados que se encuentran localizados en grandes articulaciones y las zonas más expuestas a traumatismos8.
No existe tratamiento curativo para el síndrome de Ehlers-Danlos. Hay que prevenir las consecuencias de la fragilidad cutánea, articular y vascular, evitando ejercicios y profesiones violentas, roces, excoriaciones, traumatismos, etc. En el tipo IV, se ha utilizado la terapia con DDAVP con resultados satisfactorios30.
Vasculitis
Púrpura de Schonlein-Henoch
Clásicamente, se considera que la púrpura de Schonlein-Henoch es una vasculitis reactiva, que aparece sobre todo en niños y adolescentes, y que se manifiesta clínicamente por púrpura palpable, artromialgias y alteraciones abdominales y renales. En más del 50% de casos es imposible atribuirla a ninguna causa31; sin embargo, se ha relacionado con infecciones del tracto respiratorio superior (sobre todo en la edad infantil) o con medicamentos (principalmente en la edad adulta).
Dependiendo de las series, un 50-100% de los pacientes con púrpura de Schonlein-Henoch presentan clínica gastrointestinal8; son frecuentes el dolor abdominal, los vómitos, la diarrea y la hemorragia digestiva, normalmente oculta en heces, que suele ocurrir en un 40-60% de casos32,33. Se han descrito complicaciones digestivas graves, como intususpección o perforación32. En ocasiones, la clínica gastrointestinal puede preceder a la sintomatología cutánea, simulando otras causas de dolor abdominal. Los hallazgos endoscópicos revelarán lesiones inflamatorias y purpúricas relativamente inespecíficas32.
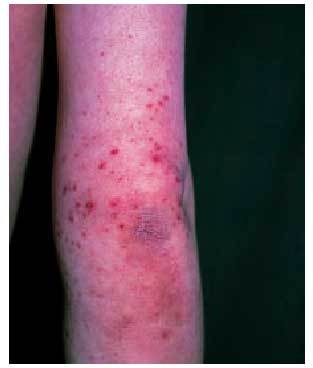
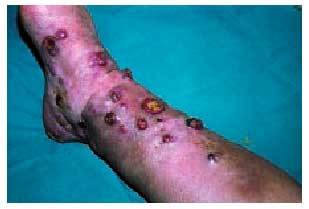
La afección cutánea suele presentarse en forma de púrpura palpable (fig. 3), principalmente en las nalgas y las extremidades inferiores, que puede evolucionar a vesiculación central, necrosis o ulceración. Esta púrpura palpable suele manifestarse poco después o concomitantemente con síntomas constitucionales, como fiebre, malestar general, cefalea o anorexia. La biopsia de una de estas lesiones revelará una vasculitis leucocitoclástica y la inmunofluorescencia directa puede objetivar el depósito de C3, IgM y/o IgA, siendo considerada clásicamente la presencia de IgA como un hallazgo específico de la enfermedad8.
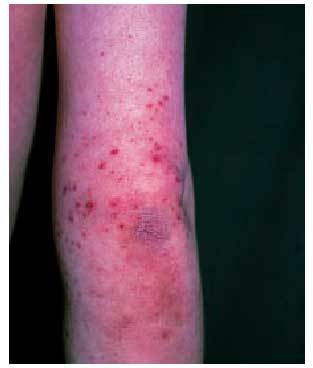
Fig. 3. Lesiones purpúricas en el brazo derecho de un niño con púrpura de Schonlein-Henoch.
Otros hallazgos clínicos frecuentes son las poliartralgias erráticas inflamatorias en muñecas, codos, rodillas, tobillos y pequeñas articulaciones de las manos, así como la afección renal en forma de síndrome nefrótico por glomerulonefritis segmentaria y focal, en un 23-49% de los casos34, y otras menos frecuentes, como edema laríngeo o escrotal, alteraciones pulmonares, oculares o síndrome cerebral seudotumoral.
El diagnóstico es clínico, aunque pueden ayudar las biopsias renal y cutánea con inmunofluorescencia directa (revelando depósitos de IgA perivasculares). Puede objetivarse en los exámenes complementarios un aumento de la VSG, leucocitosis e hipergammaglobulinemia con elevación de la IgA sérica.
El tratamiento es causal y sintomático, siendo recomendable el reposo en cama. Algunos estudios recomiendan el uso de corticoides sistémicos para atenuar los síntomas y acortar el tiempo de curación. Otros tratamientos utilizados son la azatioprina, la pentoxifilina y la dapsona.
El pronóstico es, en general, favorable (dependiendo sobre todo de las manifestaciones renales), puesto que la regla general es la resolución espontánea del cuadro en pocas semanas o meses. Sin embargo, en ese período las recurrencias no son raras35.
Papulosis atrófica maligna (enfermedad de Degos)
La papulosis atrófica maligna fue descrita inicialmente en 1941 por Köhlmeier36 y reconocida como una enfermedad específica por Degos en 194237. Es una enfermedad rara, de etiología desconocida, que se caracteriza por una vasculitis linfocítica multisistémica de pequeños vasos, produciéndose trombos que pueden afectar tanto a la piel como al tracto gastrointestinal, los ojos y el SNC. La enfermedad se presenta, generalmente, en varones jóvenes de raza caucásica, aunque han sido descritos algunos casos en niños.
Las manifestaciones cutáneas son constantes y características. Las lesiones son múltiples (fig. 4) y pasan por diferentes estadios, afectando predominantemente al tronco y las extremidades. En las primeras fases, las lesiones son pápulas inflamatorias de 2 a 5 mm de diámetro, de coloración rosada, grisácea o amarillenta, recubiertas por piel normal. A los pocos días, las pápulas comienzan a umbilicarse y aparece una zona típica ce ntral atrófica, de coloración blanco-porcelana (fig. 5), que puede estar recubierta por una costra y rodeada por un borde eritematoso. Con el tiempo, este borde desaparece y aparece una costra varioliforme. Normalmente, las lesiones son aisladas aunque pueden coalescer formando áreas atróficas o ulceradas. Las lesiones van apareciendo en forma de brotes, por lo que pueden observarse lesiones en diferentes estadios en el mismo sujeto. Estas lesiones no suelen producir sintomatología, aunque algunas veces pueden originar un ligero prurito. Se han descrito alteraciones en mucosas, afectándose predominantemente la ocular en un tercio de los pacientes38.
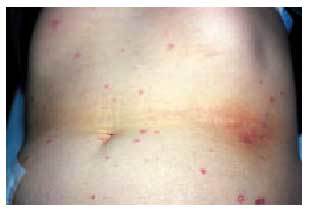
Fig. 4. Paciente con papulosis atrofiante maligna o enfermedad de Degos.
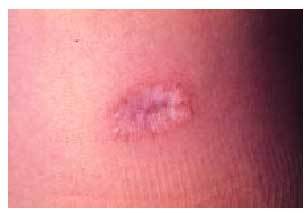
Fig. 5. Detalle de la típica lesión papulosa con área central atrófica de coloración blanco-porcelana de la enfermedad de Degos.
Normalmente, las manifestaciones cutáneas preceden a la sintomatología visceral. Cuando las manifestaciones gastrointestinales o neurológicas preceden a las lesiones cutáneas, el diagnóstico suele ser difícil de realizar.
Las lesiones gastrointestinales suelen aparecer meses o años después de las cutáneas. Según las estadísticas de los casos publicados, el 61% de los pacientes sufre una afección gastrointestinal, sobre todo en el intestino delgado. La alteración digestiva puede ser asintomática o producir síntomas de diferente gravedad, desde disfagia, vómitos, hematemesis y melenas hasta perforación intestinal que puede conducir a una peritonitis39,40. Los estudios radiológicos suelen ser normales. En la laparoscopia, la laparotomía y la tomografía computarizada de abdomen se pueden observar lesiones parcheadas avasculares que se diseminan por el intestino delgado, el colon o el estómago41. En la endoscopia pueden encontrarse lesiones de similares características a las cutáneas. La existencia de lesiones gastrointestinales se asocia a mal pronóstico.
Las alteraciones neurológicas en la enfermedad de Degos son muy variadas y se observan en el 20% de los pacientes. Pueden presentar hemiparesia, afasia, afección de los pares craneales, monoplejía y alteraciones sensoriales, además de alteraciones en el líquido cefalo rraquídeo. Los hallazgos neuropatológicos revelan infartos múltiples, con o sin hemorragias en el cerebro, el cerebelo y la médula espinal. Además, los vasos meníngeos pueden estar ocluidos42.
En la autopsia de estos pacientes se han podido observar lesiones en otros órganos (p. ej., corazón, riñones, vejiga, pulmones, pleura, hígado y páncreas). Normalmente son asintomáticas, pero en algunos casos pueden producir sintomatologías diversas.
En los hallazgos de laboratorio debemos destacar ciertas alteraciones en el estudio de coagulación, con incrementos en los valores de fibrinógeno en plasma43, elevación de la agregación plaquetaria44, disminución de la actividad fibrinolítica local y sistémica y presencia de anticuerpos anticardiolipina y anticoagulante lúpico45. También se ha descrito la posibilidad de una coagulopatía por consumo.
Las lesiones histológicas son características. La lesión típica consiste en áreas de necrosis dérmica cubierta por epidermis atrófica con hiperqueratosis leve; sin embargo, es más frecuente que en lugar de necrosis se aprecie edema, depósitos de mucina y esclerosis. Puede observarse un infiltrado linfocitario perivascular poco significativo aunque, en general, la dermis es acelular. Los vasos implicados son los de la base del área de necrosis. Las alteraciones podrían ser sutiles y traducirse en tumefacción endotelial, pero con frecuencia se encuentran trombos de fibrina que sugieren que los cambios dérmicos y epidérmicos se deben a fenómenos isquémicos.
Hasta el momento no se conoce ningún tratamiento eficaz. La administración de corticoides es discutida ya que puede incrementar el riesgo de perforación intestinal46. Los anticoagulantes, sobre todo la heparina, han sido utilizados sin aportar beneficios47. Los antiagregantes han demostrado eficacia, sobre todo en pacientes con aumento de la agregación plaquetaria44,48. También se ha demostrado eficaz el tratamiento con fibrinolíticos en algunos sujetos49. La cirugía de las lesiones gástricas no suele ser útil.
Aunque algunos pacientes han presentado una supervivencia de hasta 15 años con tan sólo lesiones cutáneas, el pronóstico es malo y los pacientes mueren en poco tiempo como consecuencia de alteraciones gastrointestinales o neurológicas.
Miscelánea
Sarcoma de Kaposi
La enfermedad se caracteriza por la proliferación de células de origen predominantemente endotelial, neoangiogénesis, infiltrado inflamatorio y edema. Fue descrito por primera vez en 1872 por Moritz Kaposi como «síndrome del sarcoma pigmentado idiopático múltiple de la piel»50; sin embargo, actualmente se sabe que el sarcoma de la descripción original es uno de los múltiples tipos y que no se encuentran limitados tan sólo al tegumento cutáneo. En su etiopatogenia, se ha demostrado el papel relevante desempeñado por el virus herpes humano-8 (VHH-8), interviniendo también otros factores genéticos y ambientales51.
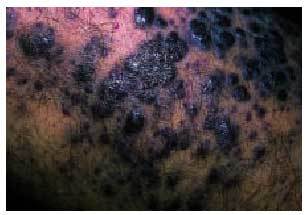
Existen 4 tipos de sarcoma de Kaposi (SK): a) SK clásico (fig. 6) que se presenta en varones, entre 40 y 70 años de edad, de origen mediterráneo y de localización predominante en las extremidades inferiores, aunque en su evolución puede extenderse a los órganos internos, incluyendo el tracto gastrointestinal; b) SK africano o endémico, que ocurre casi exclusivamente en varones de 30-40 años y niños de raza negra; c) SK en inmunodeprimidos, que se desarrolla en pacientes que han recibido trasplantes de órganos, que presentan trastornos autoinmunes o linfomas o en aquellos que han recibido tratamiento inmunodepresor, y d) SK asociado a sida o epidémico (fig. 7), que se da en sujetos de mediana edad (30-35 años) que padecen sida, siendo más frecuente en homosexuales o bisexuales y más agresivo en mujeres52. El SK epidémico es el que presenta más tendencia a la diseminación, afectándose a menudo las mucosas, el tracto gastrointestinal, los ganglios linfáticos, el hígado, el pulmón y otros órganos. La afección del tracto gastrointestinal se da en un 50-80% de pacientes con lesiones cutáneas y en casi un 100% de aquellos con afección mucosa. Puede verse afectado cualquier segmento del tracto gastrointestinal y es una de las causas más frecuentes (junto con las úlceras grastroduodenales y esofágicas) de la hemorragia intestinal aguda en pacientes con sida53. La biopsia endoscópica es recomendable para el diagnóstico, siendo la histología similar a las lesiones cutáneas.
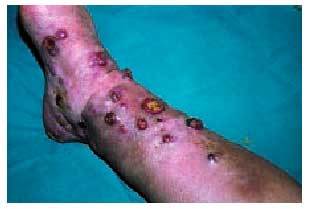
Fig. 6. Sarcoma de Kaposi clásico.
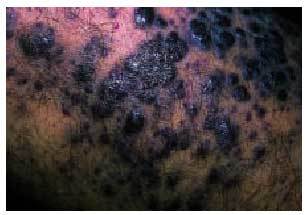
Fig. 7. Detalle de las lesiones de sarcoma de Kaposi en un paciente con sida.
Las manifestaciones cutáneas del SK consisten en un número variable de máculas, placas o nódulos de color rojo-azulado o violáceo, de distintos tamaños, aunque generalmente no superiores a unos pocos centímetros, redondeadas u ovaladas y frecuentemente rodeadas de un halo purpúrico. Generalmente son asintomáticas. Desde el punto de vista histológico, el tumor está constituido por formaciones vasculares más o menos diferenciadas con predominio de células endoteliales y acumulaciones de células fusiformes, con núcleos hipercromáticos, atípicos y/o en mitosis, entre las que se encuentran las hendiduras con hematíes en su interior.
Existen diferentes tratamientos según la forma clínica y el momento evolutivo de la enfermedad. Las lesiones clásicas y circunscritas son susceptibles de cirugía y radioterapia superficial. Cuando existen manifestaciones diseminadas o el curso evolutivo es agresivo se utilizan la radioterapia y la quimioterapia, aunque las recidivas son habituales. También se utiliza el IFN-2-alfa recombinante y, más recientemente, inhibidores de la angiogénesis, terapias hormonales, derivados del ácido retinoico e inmunomoduladores como la IL-154.
Escorbuto
En 1753, Sir James Lind describió la historia natural del escorbuto y la manera de prevenirlo con dieta rica en cítricos. En los países industrializados, más del 90% de la vitamina C deriva de las frutas y vegetales, incluyendo patatas, tomates, verduras y cítricos55. Nuestro organismo necesita 30 mg de vitamina C al día para prevenir el déficit de esta vitamina. En la actualidad, esta enfermedad es poco frecuente en los países industrializados debido a las dietas y suplementos vitamínicos que se consumen. No obstante, en determinadas circunstancias podemos encontrar esta deficiencia en pacientes con alteraciones digestivas, alcoholismo, alteraciones psicológicas, en regímenes alimenticios o por carencia de la enzima transformadora de gluconato en ascorbato.
La carencia de vitamina C modifica el colágeno y los vasos sanguíneos, además de los huesos y los dientes en crecimiento. Clínicamente, se manifiesta con alteraciones constitucionales, alteraciones cutáneas, orales, musculoesqueléticas, oftalmológicas, del sistema cardiorrespiratorio y gastrointestinales.
El cuadro suele comenzar con síntomas constitucionales, apareciendo fatiga sobre todo en las piernas y, posteriormente, confusión, somnolencia e incluso depresión56.
Las alteraciones cutáneas se inician con agrandamiento y queratosis de los folículos, a menudo en las caras laterales de los brazos. En pocas semanas la queratosis se extiende afectando a la totalidad de las extremidades superiores, los glúteos y las superficies tibiales anteriores. Cada queratosis folicular queda rodeada por un halo hemorrágico y aparecen petequias, especialmente en las extremidades inferiores, que clínicamente pueden interpretarse como una «púrpura palpable»57,58. También se han descrito hemorragias subungueales lineales «en astilla», equimosis, hematomas, edema cutáneo y alopecia, además de la mala cicatrización de las heridas59.
Las lesiones gingivales son muy frecuentes; consisten en tumefacción, reblandecimiento y vulnerabilidad de las encías, en las que se producen frecuentemente ulceraciones, hemorragias (gingivitis hemorrágica hiperplásica) y, posteriormente, necrosis, que provocarán desprendimientos dentarios. Es frecuente el aflojamiento y el desprendimiento dentario.
La anorexia es el síntoma digestivo más frecuente, aunque también puede existir hemorragia gastrointestinal. Al realizar la endoscopia, se ha observado que estas hemorragias pueden afectar cualquier zona de submucosa de esófago distal o de duodeno60,61.
Las alteraciones musculoesqueléticas se producen principalmente en las zonas sometidas a gran esfuerzo. Pueden existir hemorragias subcutáneas, musculares, articulares y periósticas, que pueden dificultar la marcha. Asimismo, se han encontrado alteraciones oculares y afección de los sistemas cardiorrespiratorios como hipertensión pulmonar, síncopes e hipotensión, que pueden causar un shock58,59.
El diagnóstico de esta enfermedad debe realizarse principalmente por la clínica, siendo el tratamiento la administración de vitamina C intravenosa (entre 500 y 1.000 mg/día).
Síndrome de Plummer-Vinson (Paterson-Brown Kelly)
El síndrome de Plummer-Vinson (o síndrome de Paterson-Brown Kelly) se describe clásicamente como la asociación de anemia por déficit de hierro con disfagia secundaria a la presencia de membranas esofágicas poscricoideas. Patterson y Brown-Kelly fueron los primeros en describir, en 1919, la asociación entre anemia y disfagia62,63. Plummer y Vinson en 1922 describieron un síndrome que denominaron «disfagia histérica», el cual carecía de obstrucción orgánica que lo justificara, pero que respondía al tratamiento con hierro64.
La mayoría de los casos, entre el 80-90%, se presentan en mujeres, sobre todo de mediana edad, pudiendo estar relacionados con pérdidas sanguíneas durante el parto o la menstruación65. Se han argumentado otras causas posibles para el déficit de hierro, como carencias nutricionales y alteraciones gástricas (p. ej., se han descrito casos de enfermedad celíaca que se manifestaron como un síndrome de Plummer-Vinson secundario a la malabsorción)66,67. Se ha asociado también a enfermedades autoinmunes.
El complejo sintomático incluye como principales manifestaciones, disfagia, anemia y glositis. La anemia hipocrómica y microcítica, debida al déficit de hierro, produce las alteraciones clásicas siguientes: coiloniquia (uñas de concavidad superior o en cuchara), aclorhidria, queilitis, sequedad de pelo y esplenomegalia. Otras alteraciones que se han asociado a este síndrome son la atrofia epidérmica, hiperqueratosis, alteraciones mucosas en lengua, hipofaringe, esófago, nariz y vagina. Las membranas esofágicas aparecen normalmente por encima del músculo cricofaríngeo y pueden producir clínica de disfagia. También se han descrito alteraciones de la motilidad esofágica secundarias al déficit de hierro que originaría una disminución de mioglobulina en el músculo esquelético provocando déficit en la motilidad68.
En estos pacientes, existe un riesgo aumentado de desarrollar tumores de cabeza y cuello, siendo los más frecuentes los carcinomas esofágicos poscricoideos y de tracto respiratorio, por lo que es importante el seguimiento evolutivo de estos pacientes.
El diagnóstico se realiza basándonos en la clínica, la confirmación analítica de anemia hipocrómica microcítica y la demostración mediante técnicas complementarias de membrana esofágica.
El tratamiento consiste en la reposición del déficit de hierro. Si la membrana esofágica produce disfagia o está muy evolucionada se debe realizar dilatación mecánica.
Neurofibromatosis tipo 1
Las neurofibromatosis (NF) son síndromes neurocutáneos hereditarios que se presentan con relativa frecuencia. Existen diferentes tipos, aunque tan sólo dos variantes generalizadas: NF tipo 1, o forma del sistema nervioso periférico, y NF tipo 2, o forma del SNC o neuroma acústico. La forma habitual o «clásica» es la NF tipo 1, que es la más frecuente y la que trataremos a continuación69. Fue descrita por el anatomopatólogo Friedrerich von Recklinghausen, en 1882, y se caracteriza por seguir un patrón de herencia autosómico dominante con gran penetrancia y expresión clínica variable. Se calcula que la prevalencia de la enfermedad varía de 1/2.190 a 1/7.800, no evidenciándose variaciones significativas de la misma en los diferentes grupos étnicos. Los estudios señalan una de las tasas más elevadas de mutación encontradas en el ser humano, lo que explica que cerca del 50% de los casos sean debidos a nuevas mutaciones. El gen de esta enfermedad fue localizado en el brazo largo del cromosoma 17(17q11,2)70,71.
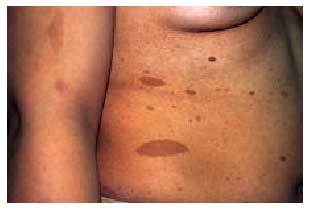
El síndrome cutáneo es, a menudo, precoz y muy evocador. Se han descrito: a) «manchas de café con leche» (fig. 8) redondeadas u ovales, de contornos regulares, en zonas de tegumento cutáneo normalmente cubiertas, que pueden estar presentes ya en el nacimiento; b) pigmentación moteada axilar e inguinal (signo de Crowe), y c) neurofibromas (fig. 9), que son tumores fláccidos o tensos, sésiles o pediculados, del color de la piel normal e indoloros, que pueden alcanzar tamaños considerables (neurofibromas plexiformes). Los criterios diagnósticos de la NF tipo 1 se exponen en la tabla 2.

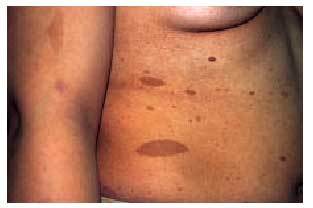
Fig. 9. Neurofibromas presentes en un paciente con neurofibromatosis tipo 1.
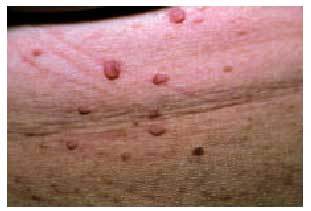
Fig. 8. Manchas de café con leche en una paciente con neurofibromatosis tipo 1.
El tracto gastrointestinal se encuentra afectado en el 25% de los casos, en forma de múltiples neurofibromas submucosos responsables de la aparición de hemorragia intestinal recurrente72,73. Habitualmente, son manifestaciones tardías de la enfermedad, pero en casos excepcionales pueden ser un signo inicial de neurofibromatosis, lo que obliga a la búsqueda de otros rasgos de neurofibromatosis tipo 1 en estos pacientes y en sus familiares74.
Al cuadro sindrómico se añaden manifestaciones neurológicas debidas al desarrollo de neurofibromas en los pares craneales (neurinoma del acústico...), psíquicas (oligofrenia...), oculares (glioma del nervio óptico, nódulos de Lisch...), endocrinas, óseas y viscerales de diversa índole.
La supervivencia en pacientes con NF tipo 1 está significativamente acortada según distintos estudios. Las causas más comunes de muerte (cáncer, infarto agudo de miocardio, accidente cerebrovascular y neumonía) son similares a las de la población general, pero aparecen en edades más tempranas70. El pronóstico también depende de la eventual malignización de los neurofibromas y la aparición de schwannomas malignos, astrocitomas, glioblastomas o fibrosarcomas, lo que provoca que sea estrictamente necesario establecer un control periódico de estas lesiones con el fin de detectarlas en un estadio precoz.
El tratamiento es sintomático y multidisciplinario. Las lesiones tumorales requieren a menudo técnicas quirúrgicas, algunas veces complicadas y con resultados decepcionantes75,76.
Síndrome del nevus azul en tetina de goma
El síndrome del nevus azul en tetina de goma fue descrito por Bean, en 1958, como un síndrome que comprende múltiples hemangiomas cavernosos. En general, se presenta esporádicamente aunque se ha descrito algún caso de herencia autosómica dominante. Suele apa recer al nacimiento o en edades tempranas, y en muy raras ocasiones lo hace en la edad adulta77.
Los tumores cutáneos se caracterizan por tener una apariencia en «tetina de goma» (formaciones angiomatosas excrecentes, lobuladas, de pocos centímetros o milímetros de diámetro, de color azul-violáceo, depresibles y elásticas). También pueden aparecer máculas puntiformes y grandes hemangiomas desfigurantes que pueden interferir en la función de la extremidad o el órgano donde asientan. Pueden ser dolorosas o asociarse a excesiva sudación78,79.
Las lesiones del tracto gastrointestinal suelen localizarse en el intestino delgado o grueso y presentan facilidad para el sangrado, por lo general microscópico, aunque en ocasiones pueden provocar francas melenas o hematoquecia. Los hemangiomas pueden también localizarse en el mesenterio, el hígado, el pulmón, los ojos y el SNC.
Las lesiones del síndrome del nevus azul en tetina de goma no regresan espontáneamente. El tratamiento es sintomático a base de suplementos de hierro y transfusiones, según la intensidad de la anemia. La coagulación controlada endoscópicamente o la cirugía del tracto gastrointestinal se reservan para los casos de hemorragia intestinal incoercible. Los hemangiomas cavernosos cutáneos pueden ser tratados con láser o agentes esclerosantes.
Síndrome de Clarke-Howel-Evans
En 1958, Howel-Evans et al publicaron el caso de dos familias inglesas con queratodermia palmoplantar difusa que fueron controladas temporalmente, y constataron que, aproximadamente el 70% de los afectados fallecieron por un carcinoma de esófago80. Actualmente, el síndrome de Clarke-Howel-Evans se define como la asociación de queratodermia palmoplantar y cáncer de esófago81. La presencia de hiperqueratosis folicular, quistes triquilemales axilares, adelgazamiento de las pestañas y leucoplaquia oral puede completar el cuadro cutaneomucoso de este síndrome.
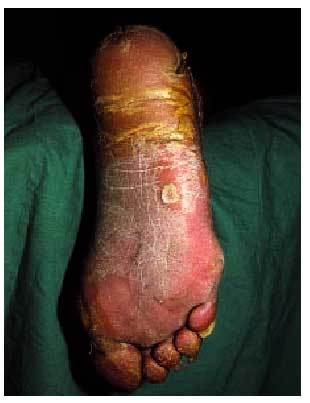
La queratodermia (o aumento de la capa córnea) palmoplantar (fig. 10) presente en este síndrome es difícilmente distinguible de las queratodermias benignas aunque, en general, es de aparición más tardía, sobrepasa la localización palmoplantar (transgrediens), tiene un curso progresivo (progrediens) y suele preceder al desarrollo del carcinoma esofágico hasta en dos décadas. Este último hecho hace necesaria la realización de exploraciones periódicas en pacientes con queratodermias y antecedentes de neoformaciones esofágicas80-82.
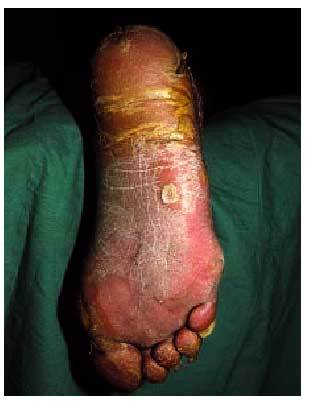
Fig. 10. Hiperqueratosis palmoplantar.