




las manifestaciones comunes de ambas y sugerencias de cuidado, así como el tratamiento que es sintomático con seguimiento multidisciplinario.
Las neurofibromatosis son un grupo de enfermedades genéticas multisistémicas, heredadas de forma autosómica dominante con implicación patogénica de la piel, el sistema nervioso, los ojos, los huesos y el sistema endocrino, y con un amplio espectro de hamartomas, tumores malignos y alteraciones congénitas.
Las clasificaciones han sido variadas, desde los 7 tipos de Riccardi (clásica, acústica, mixta, inclasificable, segmentaria, sólo manchas «café con leche», sólo neurofibromas), pasando por las 4 formas de Houson (clásica, acústica, segmentaria, manchas «café con leche» familiares) hasta la clasificación que aquí vamos a emplear según la cual sólo se consideran 2 tipos: neurofibromatosis tipo 1 (NF-1) y neurofibromatosis tipo 2 (NF-2) con una variante de NF-1 que sería la neurofibromatosis segmentaria.
Neurofibromatosis tipo 1 o enfermedad de Von Recklinghausen
Es la más frecuente de todas, hasta el 97% de todos los tipos de NF diagnosticadas. Descrita en 1882 por el anatomopatólogo Friedreich von Recklinghausen, afecta a una de cada 3.000 personas, y se ha calculado que cada médico se encontrará por lo menos 2 casos en su vida profesional. También se llama NF periférica, clásica o tipo 1 que es el término actual.
Es una enfermedad autosómica dominante, aunque hasta en el 50% de los casos es de aparición esporádica por mutación del gen (es la segunda tasa más alta de mutación genética tras la acondroplasia). El gen de la NF-1 está localizado en el cromosoma 17 q 11.2, caracterizado y secuenciado desde 1990; es un gen largo, de 350 kb de ADN conteniendo 60 exones, y codifica una proteína llamada neurofibromina que pertenece a una familia de proteínas GAP que sirve como regulador negativo del oncogén ras. Esto es, el gen de la NF-1 es un gen supresor cuyo defecto provoca la aparición de tumores y hamartomas al dejar de ejercer esa supresión tumoral.
Hasta 1999 se habían constatado más de 240 posibles mutaciones en el gen de la NF-1 (deleciones, inserciones, translocaciones, etc.) algunas de las cuales no pueden determinarse por los tests comerciales de diagnóstico disponibles, encaminados a detectar alteraciones en la proteína final formada codificada por ese gen. Las mutaciones, en un 90%, derivan del cromosoma paterno aunque las grandes deleciones son de origen materno. La enfermedad tiene una penetrancia del 100%, aunque con un fenotipo muy variable sin poderse aún predecir qué mutación provocará determinado fenotipo o si éste se expresará completamente en ese individuo, lo cual es un auténtico escollo para decidir actitudes terapeúticas prenatales.
Clínica
En 1987, el Instituto Nacional de la Salud americano estableció los actuales criterios de diagnóstico que siguen siendo fundamentalmente clínicos y que podemos ver en la tabla 1. Son útiles en un 94% a la edad de 6 años, o desde otra valoración, si a los 5 años no cumple estos criterios, podemos asegurar que no va a desarrollar la enfermedad con un 95% de seguridad. Sin embargo, en los niños de 2-4 años estos criterios bajan su porcentaje de utilidad de forma considerable y últimamente se está dando una importante relevancia a otra serie de manifestaciones clínicas, sobre todo en la primera infancia: macrocefalia, estatura corta, imágenes brillantes en la resonancia magnética nuclear cerebral, facies peculiar con implantación auricular baja o displasia vertebral.

De todas formas, veamos las características clínicas, tanto en la piel como en otros órganos, que aparecen en estos pacientes, estén o no incluidos en los criterios diagnósticos.
Manchas «café con leche»
Constituyen la característica más frecuente de aparición, hasta en un 99% de los pacientes, así como la más temprana, ya que se observa en ocasiones desde el nacimiento o en la infancia temprana aumentando en número y tamaño con la edad. Son un criterio diagnóstico si se aprecian 6 o más de más de 5 mm en el individuo prepuberal o 15 mm si ha desarrollado ya la pubertad.
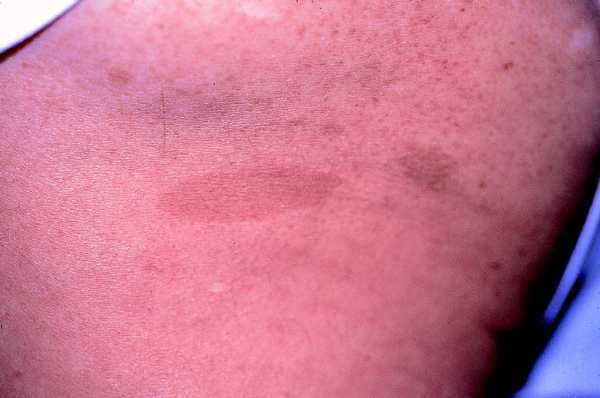
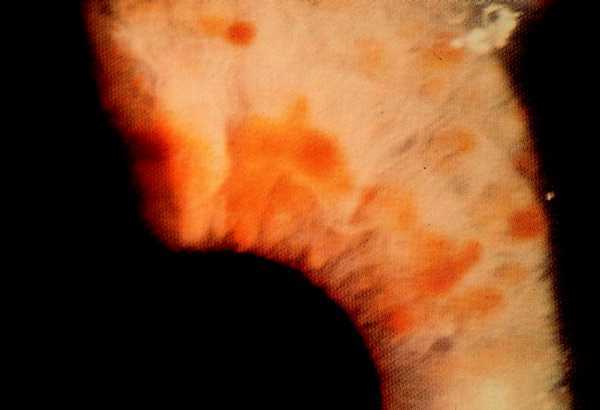
Las formas clínicas son muy variadas, con manchas que van desde el marrón claro al oscuro, de color homogéneo, borde suave y redondeado, afectando sobre todo al tronco y las extremidades ya que son raras en la cara, el cuero cabelludo, las palmas y las plantas (fig. 1).
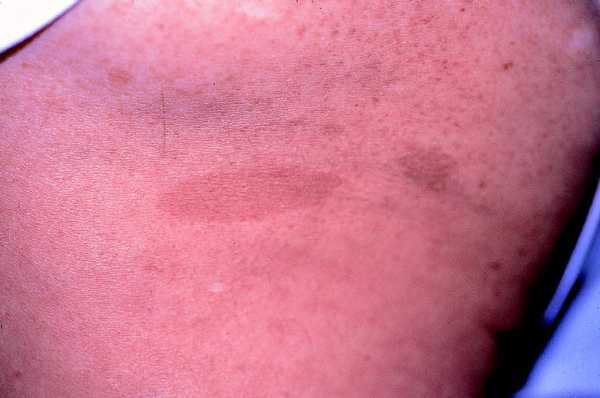
Fig. 1. Mancha «café con leche», marrón clara uniforme, borde liso.
Aunque son un dato casi constante, no son patognomónicas de la NF ya que se aprecian en otras enfermedades como la esclerosis tuberosa, el síndrome de McCune Albright, etc.
La duda radica en caso de un paciente, por ejemplo, recién nacido, con 3 o 4 manchas «café con leche» ya que la familia, a veces con otros casos ya de NF, querrá saber si va a desarrollar la enfermedad; a día de hoy sólo se podrán dar «impresiones» clínicas que se pueden apoyar en datos como macrocefalia, hipertelorismo, datos de resonancia magnética nuclear (RMN), etc. Algún estudio cifra en un 59% las posibilidades de desarrollar una NF-1 si en un recién nacido vemos más de 6 manchas «café con leche».
Neurofibromas
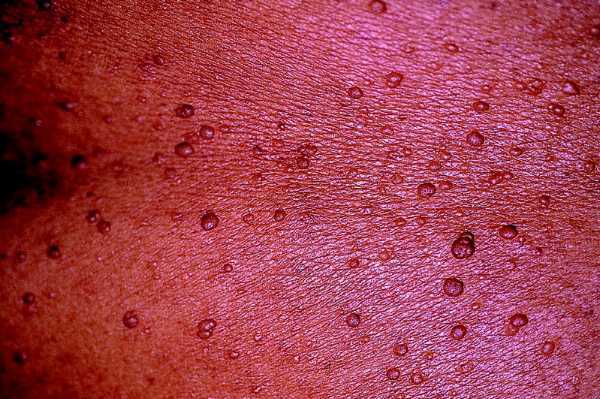
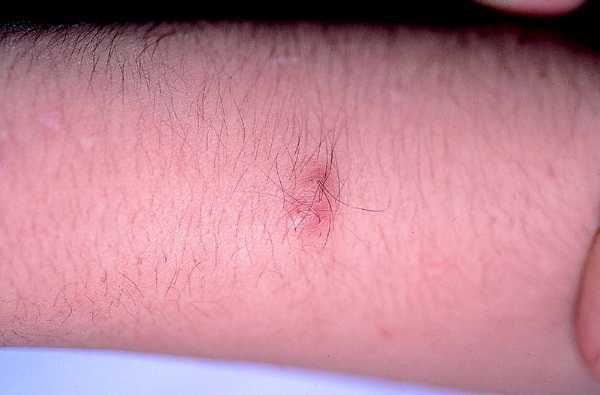
Son los tumores observados con mayor frecuencia aunque son de aparición más tardía, cerca de la pubertad y también aumentan en número y tamaño con la edad. Se ven en el 80% de los pacientes en la tercera década de la vida y pueden ser de dos tipos: los más numerosos o dérmicos de consistencia blanda, sésiles o pediculados, invaginables en ocasiones, más o menos pruriginosos, de tamaño muy variable, que suele aumentar en los embarazos, asociados a otros más profundos sin apenas relieve que siguen trayectos nerviosos (figs. 2 y 3).
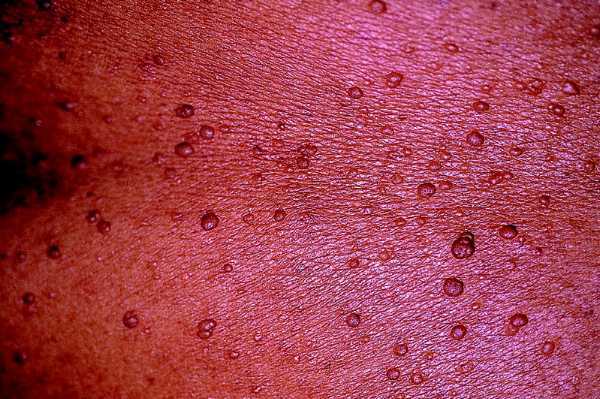
Fig. 2. Neurofibroma dérmico, pediculados, múltiples, en tronco.
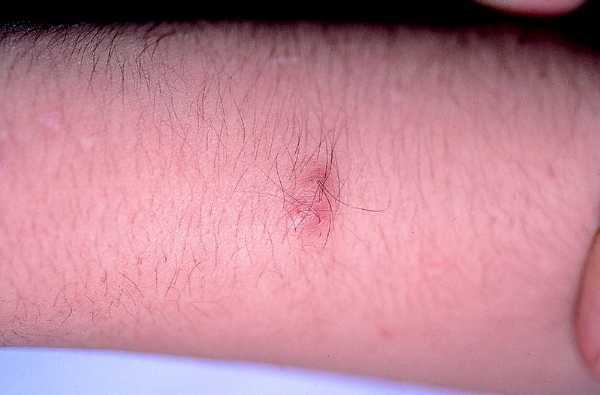
Fig. 3. Neurofibroma profundo, palpable, doloroso.
En ocasiones (15%) aparece un neurofibroma muy característico de la NF-1, el llamado neurofibroma plexi forme, a veces de varios kilogramos de peso, que pue de comprimir estructuras vecinas si asienta en el bra zo, el cuello, etc. y que presenta una mayor tasa de malignización que el resto de neurofibromas, por lo que debe considerarse su extirpación profiláctica si es posible.
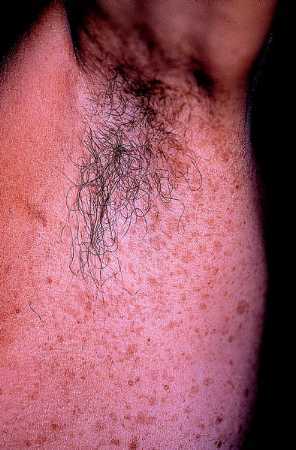
Efélides axilares
Es el llamado signo de Crowe y aparece en un 80% de los casos siendo un dato casi exclusivo de la NF-1 ya que no lo presentan las otras variedades de NF. Estas pecas, que suelen aparecer en la infancia tardía o en la pubertad, pueden afectar a las ingles u otros pliegues pero con mucha menor frecuencia.
Otros signos cutáneos que se observan en la NF-1 son la hipertrofia de papilas en mucosas hasta en un 32%, la presencia de xantogranulomas juveniles que se han relacionado con una mayor incidencia de leucemia mielomonocítica juvenil, así como la sintomatología pruriginosa generalizada en concordancia con la mayor cantidad de mastocitos que hay en la piel y los neurofibromas de estos pacientes.
Manifestaciones oculares
La clínica extracutánea no se relaciona con el grado de intensidad de los signos dermatológicos, por lo que puede ayudar a diagnosticar casos poco evidentes a nivel dermatológico y constituyendo algunos de los procesos auténticos criterios diagnósticos.
Nódulos de Lisch. Son el signo más típico, y aparecen durante la adolescencia y en la vida adulta (el 25% los tienen en la infancia tardía, el 50% a los 29 años, y casi el 100% a los 60 años). Son hamartomas pigmentarios del iris que sólo producen síntomas si distorsionan la pupila, y para apreciarlos debe existir la exploración con lámpara de hendidura. Es un criterio muy útil en pacientes o familiares asintomáticos para investigar la rama familiar de herencia.
Gliomas ópticos (astrocitomas pilocíticos). Son el otro dato encontrado a nivel ocular, hasta en un 15% de los casos y a una edad tan temprana como los 9-10 años. El diagnóstico se apoya en técnicas de imagen (RMN) ya que la clínica sólo se produce si causan proptosis, disminución de agudeza visual, pero esto sólo pasa en el 30% de los gliomas. El tratamiento va desde la cirugía a la radioterapia valorando las secuelas que cada actitud va a provocar.
Manifestaciones óseas
Se descubren alteraciones óseas en un 30% de los casos siendo el espectro clínico muy amplio, desde una macrocefalia precoz, una estatura corta, hasta una patología ósea tipo escoliosis (10-20%) leve o más intensa con posible displasia vertebral acompañante que afecta incluso a niños.
También se producen seudoartrosis congénitas de tibia y radio, displasias o agenesias del esfenoides, prognatismo mandibular o erosiones óseas por tumores neu rales.
Manifestaciones neurológicas
Constituye un aspecto importante de la enfermedad, ya que marcará el desarrollo social del individuo.
Desde la primera infancia y sobre todo posteriormente aparecen en la RMN en T2 (no en la tomografía axial computarizada [TAC]), en el 60% de los pacientes, unas lesiones localizadas en los ganglios basales y el cerebelo que se han definido como «objetos brillantes no identificados» y que pueden corresponder a pequeños astrocitomas, con una correlación clínica muy discutida, ya que existen opiniones que los asocian a retraso mental si son numerosos, pero otros dudan de su trascendencia clínica. Lo que sí es evidente es que en la NF-1 hay un mayor porcentaje de individuos con dificultad en el aprendizaje, con niveles intelectuales pobres respecto a la población normal (un 40% tiene dificultad en el razonamiento, en la pronunciación, y un 10% son retrasados mentales, frente al 2% de la población normal).
La epilepsia no es frecuente, salvo existencia de tumores gliales, y otro dato que sí observamos es la hidrocefalia a edad juvenil, generalmente de causa obstructiva y que obliga a descartar procesos neoformativos; esta hidrocefalia puede influir en el desarrollo de macrocefalia en estos individuos, sobre todo si es de aparición temprana.
Otros
En la NF-1 también podemos encontrar clínica diges tiva, como estreñimiento por alteración del plexo nervioso o neoformaciones neurales digestivas, o endo crina con una mayor frecuencia de hipertensión arte rial ya sea por desarrollo de feocromocitomas o por obstrucción de la arteria renal, y también pubertad precoz.
El riesgo más elevado es la aparición de malignización en las lesiones, siendo lo más frecuente el neurofibrosarcoma que deberemos sospechar cuando alguna lesión, sobre todo el neurofibroma plexiforme, cambia bruscamente de tamaño o altera su sintomatología dolorosa. También pueden observarse rabdomiosarcomas o tumores carcinoides.
En los últimos años se ha relacionado la presencia de NF-1 con la leucemia mielomonocítica juvenil (con un riesgo hasta 300 veces superior), sobre todo si presentan a nivel cutáneo xantogranulomas juveniles, todo ello relacionado a nivel cromosómico por una proximidad de los genes implicados.
Seguimiento, tratamiento y pruebas genéticas
Evidentemente el primer punto debe ser un diagnóstico preciso y la previsión de complicaciones y, por ello, el seguimiento es multidisciplinario con participación de pediatras, oftalmólogos, médico de familia, dermatólogo, genetista.
El diagnóstico debe ser corroborado por sus manifes taciones clínicas, los signos exploratorios cutáneos, óseos, neurológicos y oftalmológicos, tanto en el paciente como en sus familiares de primer grado. Anualmente se controlará el desarrollo ocular y su agudeza visual, la posibilidad de hipertensión arterial (HTA), escoliosis y desarrollo psicomotriz escolar. La RMN constituye un elemento importante para el seguimiento de tumores neurales internos, óseos y oculares, aunque algunos dudan de su utilidad en exploraciones rutinarias asintomáticas.
El punto más importante, y aún sin solución definitiva, es el posible diagnóstico genético, tanto en el paciente sospechoso como con diagnóstico prenatal, sobre todo si alguno de los padres presenta la enfermedad puesto que la posibilidad de transmitirlo es del 50%. Conocemos que la alteración se encuentra en el brazo largo del cromosoma 17, pero también sabemos que hay más de 240 posibles mutaciones y con un importante matiz: no conocemos qué fenotipo desarrollará una determinada mutación, esto es, a día de hoy, con las técnicas y las condiciones familiares necesarias, podemos llegar a detectar en un 80-90% si tienen una alteración genética, pero no podemos prever el fenotipo de la enfermedad en ese paciente concreto, que puede variar desde unas pocas manchas y algún neurofibroma hasta el desarrollo completo de la enfermedad con todos los signos y síntomas que hemos expuesto.
Hoy día, disponemos de lo siguiente: las diversas mutaciones del gen se traducen en la producción alterada de una proteína (truncada), que es lo que se se puede detectar. Esto permite el diagnóstico en un 70% de los casos que llega al 90% si tenemos la mutación localizada, aunque para ello necesitamos más de dos familiares afectados. La situación ideal es confirmar este diagnóstico por secuenciación del ADN u otra técnica desarrollada en servicios de genética, como la hibridación
in situ.
Todavía hay camino por recorrer ya que, como hemos visto, no es una técnica fácil y tampoco es útil como cribado en la población normal.
Así pues, lo que sí debemos ofrecer es el consejo genético (un 50% de la descendencia lo va a padecer), la posibilidad de un estudio familiar en un servicio de genética, el seguimiento y diagnóstico precoz de las posibles complicaciones para una terapia paliativa, y la oferta de información en la «red informática» con asociaciones de afectados (www.nf.org).
Neurofibromatosis segmentaria
Considerada por algunos autores como una variante de la anterior, se han descrito unos 100 casos en la bibliografía. Se trata de la aparición de manchas «café con leche» asociadas a neurofibromas (o incluso estos últimos solos) en un único territorio corporal (rama mandibular, [fig. 4], extremidad, etc.).
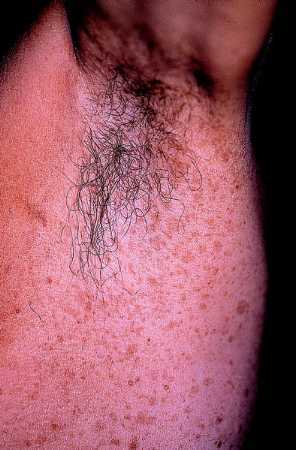
Fig. 4. Efélides de la axila, múltiples y pequeñas. Signo de Crowe.
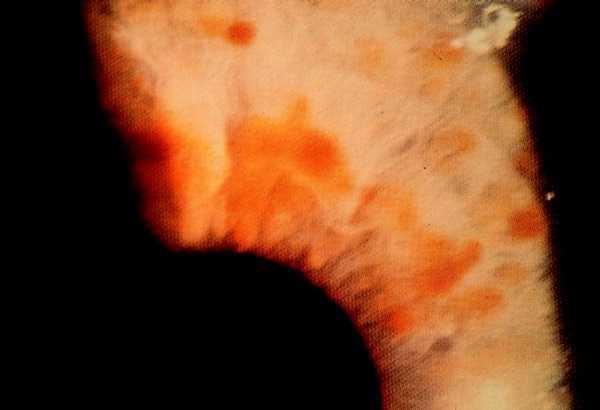
Fig. 5. Nódulos de Lisch, hamartomas de iris (lámpara de hendidura).
Patogénicamente se ha observado una anormalidad en una copia del gen de la NF-1 en unas células, respetando otras, y se cree que es debido a una deleción en este gen con una mutación poscigótica, esto es, durante la embriogénesis, produciendo este fenotipo segmentario. Es el llamado mosaicismo genético.
La duda estriba en si esta enfermedad puede conllevar descendientes con NF-1 y, aunque no se han descrito casos, en el 20% de las NF segmentarias hay algún signo sistémico, aunque un mosaicismo no debe transmitirse genéticamente. Los tests genéticos en esta variante no tienen utilidad.
Neurofibromatosis tipo 2
Es una enfermedad bastante distinta de la NF-1 y mucho más infrecuente, estimada en una de cada 50.000 personas.
Es de carácter autosómico dominante, y el gen alterado se halla en el cromosoma 22 (22q12, clonado en 1993), que es también un gen supresor y que codifica para formar una proteína llamada merlina.
A nivel cutáneo, aunque con bastante poca frecuencia, podemos encontrar alguna mancha café con leche y algún schwannoma (que clínicamente son como los neurofibromas), pero la sintomatología más importante y frecuente es la auditiva y la tumoral neural, como vemos en los criterios que definen la NF-2 (tabla 2), antes llamada neurofibromatosis central o acústica.

La importancia radica con frecuencia en los schwannomas vestibulares (antiguamente denominados neuromas o neurinomas del acústico) que pueden estar quiescentes durante años y que con estricto control de su posi ble crecimiento (con RMN con galodinio que es más sensible), puede no ser necesaria su extirpación, pero ésta debe realizarse si comienza a crecer ya que la ci rugía del schwannoma, si queda limitada a la porción vestibular del VIII par, puede conservar la funciona lidad.
Los schwannomas pueden afectar tanto los pares cra neales como los nervios periféricos, tanto en el plexo neural como en el intracraneal o el intramedular, hecho importante, ya que es la compresión en su crecimiento lo que suele motivar la cirugía, y que detectamos por técnicas de imagen, sobre todo la RMN.
La clínica suele aparecer en la pubertad con disminución de agudeza auditiva, vértigos, zumbidos, o en ocasiones dolor en la masa muscular inervada por el nervio afectado, o bien con disminución de agudeza visual por desarrollo de cataratas subcapsulares, y todo ello también puede aparecer en el adulto, por lo que la vigilancia será crónica o hasta una edad de 45-50 años.
La inteligencia de los pacientes es normal y los tests genéticos también pueden realizarse como en la NF-1, aunque con las mismas premisas y con algo menos de éxito (un 65% de los casos).
Bibliografía general
Hager C, Cohen P, Tschen J. Segmental neurofibromatosis: case report and review. J Am Acad dermatol 1997; 37: 864-869.
Karnes P. Neurofibromatosis: a common neurocutaneous disorder. Mayo Clin Proc 1998; 73: 1071-1076.
Landau M, Krafchick B. The diagnostic value of café-au-lait macules. J Am Acad Dermatol 1999; 40: 877-890.
Mautner V, Lindenau M, Baser M, Kluwe L, Gottschalk J. Skin abnormalities in NF-2. Arch Dermatol 1997; 133: 1539-1543.
North K. Neurofibromatosis 1 in chilhood. Semin Pediatr Neurol 1998; 5: 231-242.
Rasmussen S, Friedman JM. NF-1 gene and neurofibromatosis 1. Am J Epidemiol 2000; 151: 33-40.
Roos K, Muckway M. Neurofibromatosis. Dermatol Clin 1995; 1: 105-111.