


La dermatosis ampollar por IgA lineal del adulto es un desorden vesículo-ampolloso autoinmune, poco frecuente, caracterizado por la presencia de anticuerpos de IgA lineal en la membrana basal dirigidos contra diversas proteínas en la unión dermoepidérmica, entre ellas las proteínas de 97, 120, 200, 250 y 285 kD. El cuadro clínico es heterogéneo y puede llegar a confundirse con otras entidades como la dermatitis herpetiforme o el penfigoide ampolloso. Típicamente, la dermatosis por IgA lineal se manifiesta con vesículas o ampollas subepidérmicas de contenido seroso o hemorrágico sobre piel normal, eritematosa o placas de urticaria. Suele presentarse a partir de la cuarta década de vida y su etiología es mayormente idiopática. Se ha asociado con el uso de algunos medicamentos como vancomicina; sin embargo, estudios recientes cuestionan la veracidad de esta relación. Histológicamente, se observan ampollas subepidérmicas con abundantes neutrófilos y algunos eosinófilos; sin embargo, el diagnóstico definitivo se realiza mediante inmunofluorescencia directa, demostrando la presencia de depósitos de IgA lineal en la membrana basal. El tratamiento principal para este padecimiento son las sulfonas. A continuación se expone el caso de una paciente femenina de 72 años con diagnóstico de dermatosis por depósitos de IgA lineal, así como su abordaje y tratamiento.
Linear IgA bullous dermatosis is an uncommon, vesicle-bullous autoimmune disease characterized by the presence of IgA antibodies in a linear disposition at the basement membrane zone, which are directed against proteins at the dermoepidermal junction, such as 97 kD, 120 kD, 200 kD, 250 kD and 285 kD proteins. The clinical manifestations are heterogeneous and can be confused with other bullous disorders such as dermatitis herpetiformis or bullous pemphigoid. Typically, linear IgA dermatosis consists on subepidermal vesicles or bullae of serous or hemorrhagic content on normal, erythematous, or urticarial skin. It presents commonly after the fourth decade of life. The etiology is usually idiopathic, though it has been associated with the use of some drugs like vancomycin. Histologically, subepidermal bullae with many neutrophils and some eosinophils can be seen; though, the definite diagnosis is made by a direct immunofluorescence test, demonstrating the presence of linear IgA antibodies at the basement membrane zone. The first-choice treatments for this disease are sulfones. We present the case of a 72 year-old female with linear IgA bullous dermatosis, its assessment and treatment.
Introducción
La dermatosis ampollar por IgA lineal del adulto es una enfermedad autoinmune vesículo-ampollosa caracterizada por ampollas subepidérmicas tensas, de contenido sérico o hemorrágico sobre piel sana, placas eritematosas y/o placas urticariformes.1 El inicio suele ser agudo o gradual y se asocia a prurito en un 90% de los casos, el cual puede preceder o acompañar a las lesiones típicas de la enfermedad.2,3 La topografía más comúnmente afectada es tórax y extremidades (sobre todo las inferiores),4 sin embargo, hasta un 50% a 70% de los pacientes puede presentar involucro de las mucosas oral u ocular, y menos comúnmente de las superficies extensoras como codos y rodillas.2,5 La edad de inicio es variable, generalmente se presenta a partir de la cuarta década de vida, con un pico de incidencia a partir de los 60 años, siendo la incidencia similar en ambos sexos o ligeramente mayor en mujeres que hombres.1,4 A continuación se presenta el caso de una mujer de 72 años de edad, diagnosticada con dermatosis ampollar por IgA lineal, haciendo énfasis en el diagnóstico y tratamiento.
Presentación del caso
Femenino de 72 años de edad, con antecedente de diabetes mellitus tipo 2, de 15 años de duración, sin antecedentes familiares de importancia; inició un mes previo a consulta con dermatosis localizada en codo, antebrazo y mano del lado derecho, caracterizada por placas eritematosas pruriginosas que después evolucionaron a ampollas de contenido seroso, diseminándose a antebrazo y mano del lado izquierdo, pliegues interdigitales de ambas manos, ambas extremidades inferiores (piernas, fosa poplítea y parte posterior de los muslos) y glúteos. La paciente presentaba prurito intenso en todas las áreas mencionadas. La enfermedad progresó y un mes después del inicio se presentó a consulta de Dermatología. A la exploración física se observaron ampollas y costras hemáticas, en las zonas antes mencionadas (Figura 1). Se solicitó biometría hemática, química sanguínea y pruebas de función hepática, estudios que resultaron normales, observándose únicamente elevación de la glucosa en ayunas. La biopsia de piel perilesional estudiada con tinción de hematoxilina & eosina, mostró una ampolla subepidérmica con infiltrado inflamatorio mixto de neutrófilos y eosinófilos. El estudio de inmunofluorescencia directa demostró la presencia de depósitos lineales de IgA, C3 y en menor intensidad IgG en la membrana basal epidérmica (Figura 2). Se realizó el diagnóstico de dermatosis ampollar por IgA lineal del adulto y se inició tratamiento con sulfona 100 mg/día, hidroxicina 25 mg/día, clobetasol 0.05% crema una vez al día y fomentos de polvo de sulfato de aluminio diluido en 1:1 000 CCL de agua dos veces al día. Se revaloró a la semana observándose mejoría parcial, por lo que se aumentó la dosis de sulfona a 200 mg al día. Durante su evolución se descartó anemia hemolítica mediante estudios seriados de biometría hemática. La paciente evolucionó satisfactoriamente durante el primer mes, por lo que se suspendieron los tratamientos tópicos y la hidroxicina oral. Cuatro meses después, la paciente presentó curación de todas las lesiones y se decidió suspender el tratamiento indicado. Actualmente, la paciente permanece libre de lesiones desde hace año y medio.
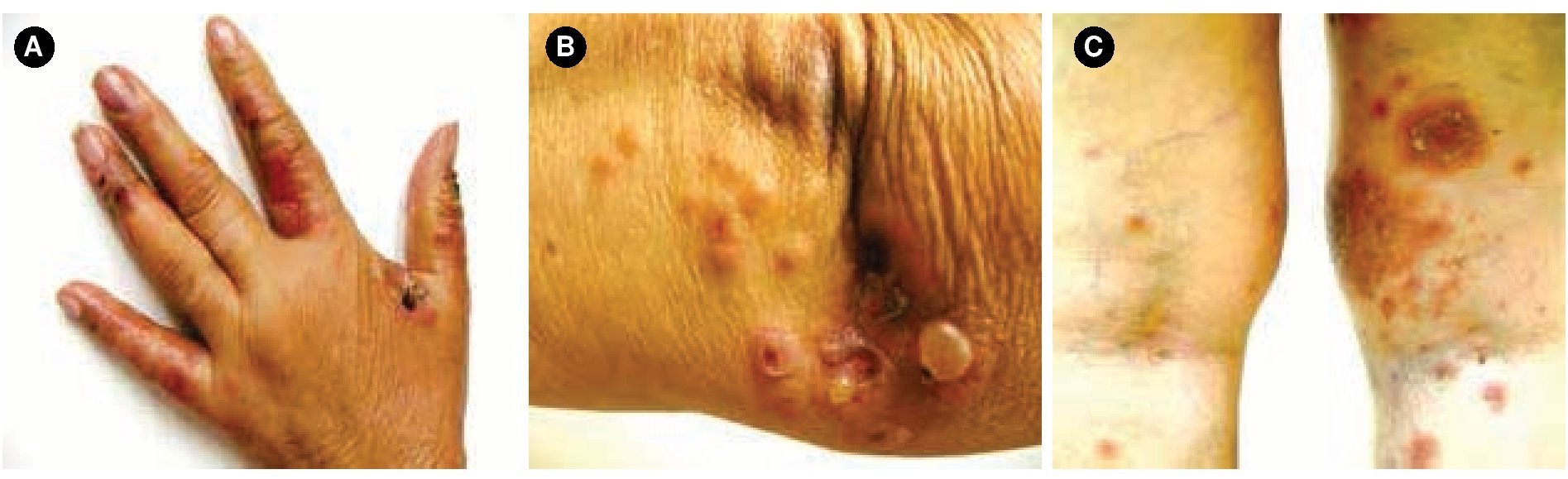
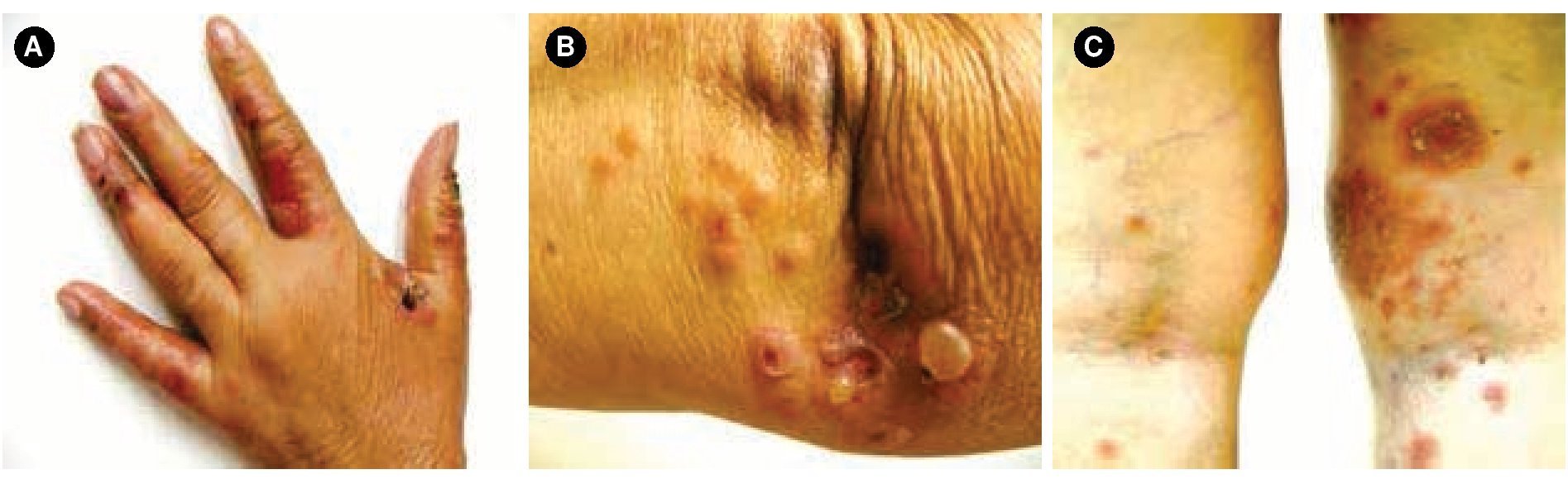
Figura 1.A) Se observa dermatosis en dedos, B) codos y, C) parte posterior de miembros inferiores, caracterizada por ampollas tensas, costras hemáticas, y pápulas/placas eritematosas.
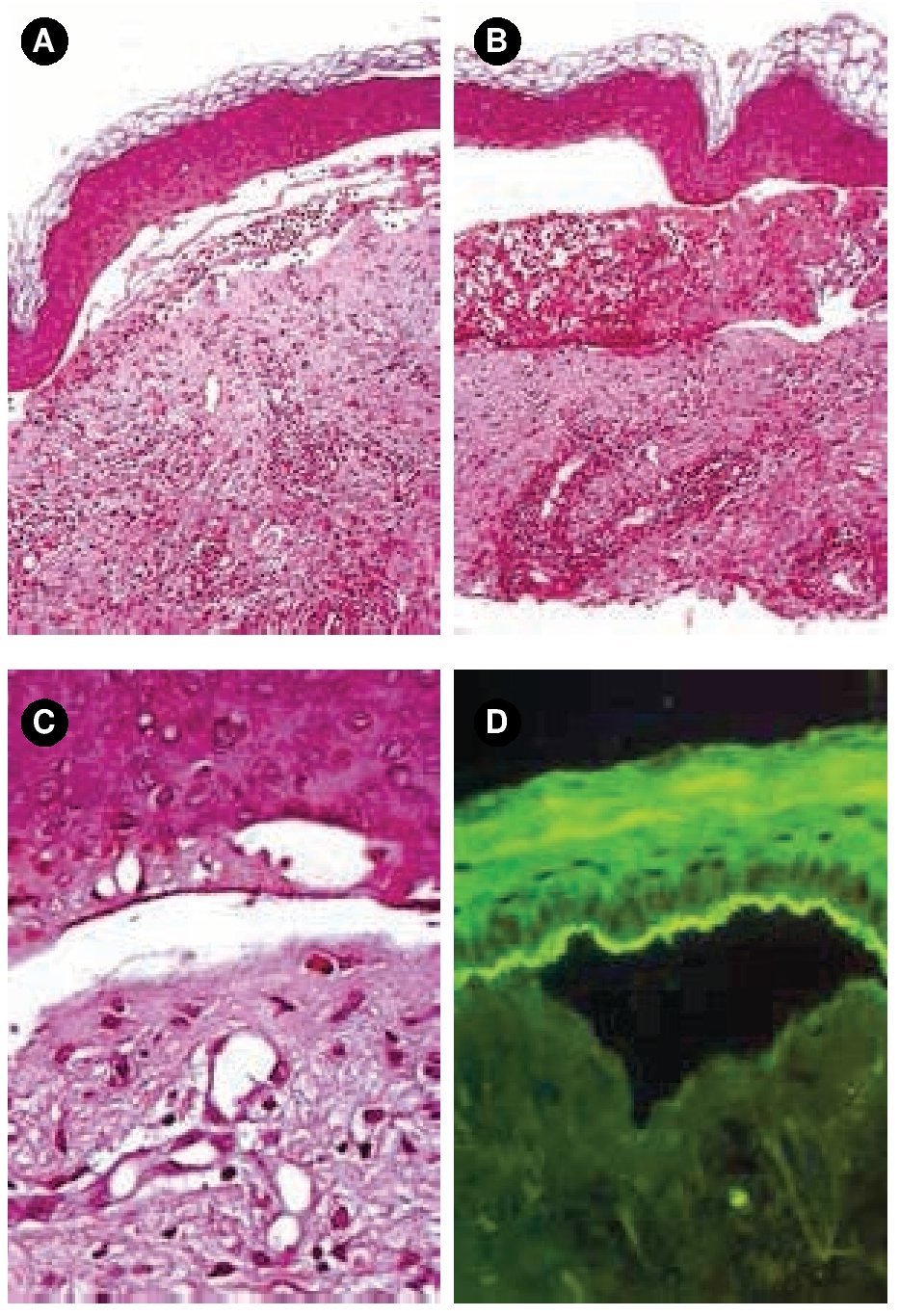
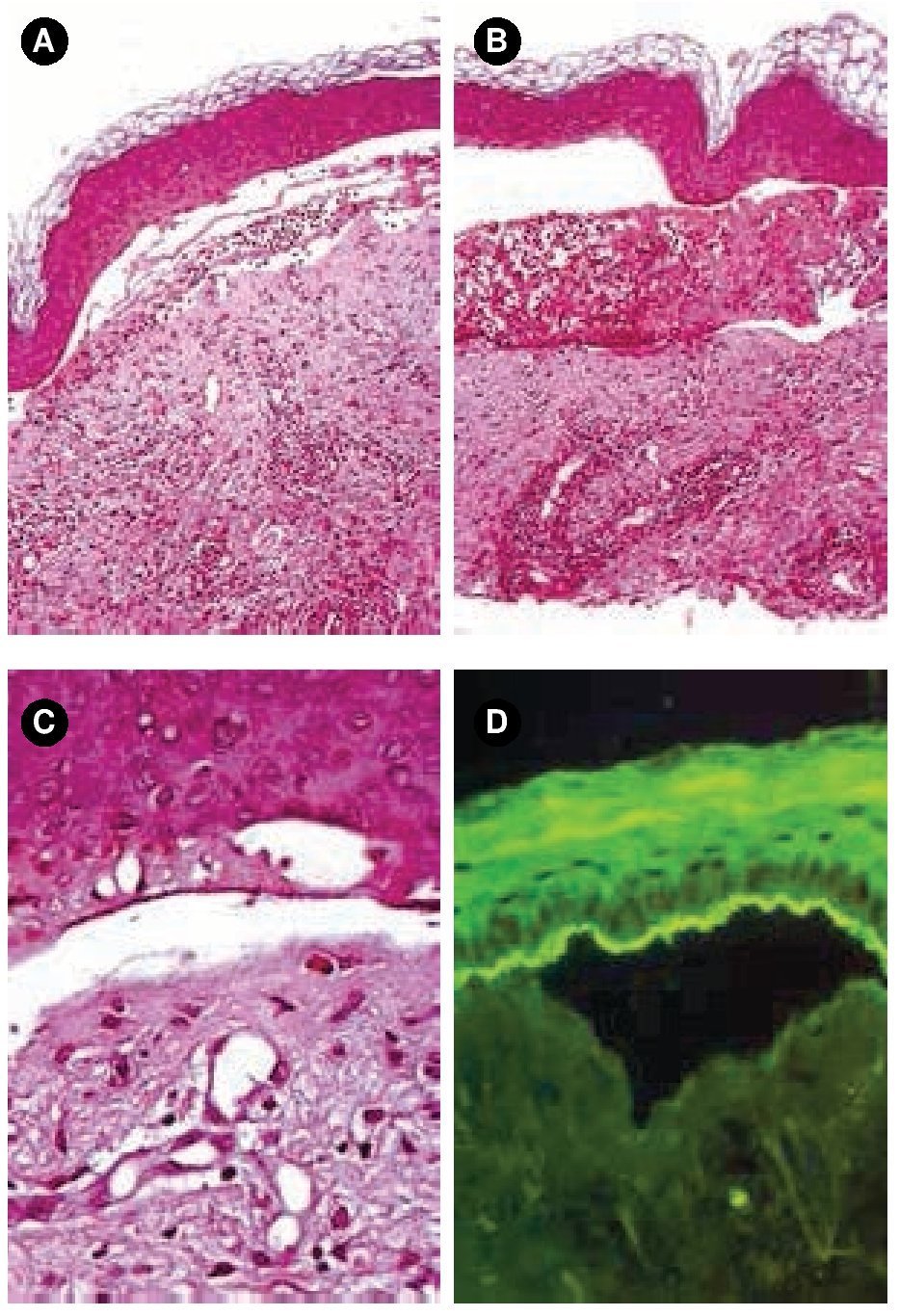
Figura 2.A) y B) Tinción con hematoxilina & eosina, 10x. C) Tinción con hematoxilina & eosina, 40x. Ampolla subepidérmica acompañada de infiltrado inflamatorio mixto en la dermis papilar. D) Patrón lineal con IgA en techo de la ampolla.
Discusión
La dermatosis por IgA lineal es una enfermedad ampollar subepidérmica poco común, de etiología desconocida, caracterizada por depósitos lineales y homogéneos de IgA en la membrana basal epidérmica. Existen dos variantes clínicas según la edad de aparición. La primera es la dermatosis ampollar crónica de la infancia, cuya edad de aparición es inferior a los cinco años, afecta por igual a ambos sexos y presenta lesiones típicas en cara, área peribucal y periné; esta entidad es autolimitada, y suele remitir a partir de los dos años del inicio.4,7,8 La segunda forma es la más común y corresponde desde el punto de vista clínico y de laboratorio, con la observada en el caso presentado. Nuestra paciente sufre de dermatosis ampollar por IgA lineal del adulto, cuyo inicio ocurre después de la pubertad, con un pico de incidencia después de los 60 años; la afectación suele ser igual o ligeramente mayor en mujeres que hombres y se manifiesta principalmente en el tronco y la mucosa oral; las lesiones suelen ser más numerosas y dispersas, y tiene una evolución impredecible que va desde remisiones espontáneas (más comúnmente), hasta una forma persistente o incluso en brotes que son menos severos que el ataque inicial.1,4 Anteriormente, la dermatosis por IgA lineal se consideraba como un subgrupo de la dermatitis herpetiforme; sin embargo, hoy en día se sabe que son entidades distintas, ya que en la primera no existe enteropatía sensible a gluten ni anticuerpos IgA antiendomisio, aunque sí se ha reportado la presencia de anticuerpos antigliadina.9
La mayoría de los casos de enfermedad por IgA lineal son idiopáticos, tal como sucedió en nuestra paciente, sin embargo, se ha establecido una relación causal con patologías sistémicas como sarcoidosis, psoriasis, colitis ulcerativa, artritis reumatoide, linfoma, tumores malignos, insuficiencia renal crónica, entre otras. También se ha reportado su aparición tras la administración de fármacos como vancomicina, captopril, somatostatina, atorvastatina, furosemida, penicilina, ceftriaxona, metronidazol, trimetoprim-sulfametoxazol, amiodarona, litio, fenitoína y algunos antiinflamatorios no esteroideos.9,10 Un estudio publicado en junio 2012 por Giulio Fortuna y Salas-Alanís y colaboradores,11 cuestionan la veracidad de esta relación. Se trata de un estudio retrospectivo de revisión, que analiza 46 artículos de enfermedad por IgA lineal inducida por medicamentos, registrados en el sistema de PubMed de 1980 al 2010. De un total de 52 casos, se observó remisión sólo en 27 de los pacientes (51.9%) tras suspender el medicamento. Se cree que la ausencia de mejoría en el resto de los pacientes podría deberse a una reacción inmunológica amplificada y autosostenida. De los 52 casos en cuestión, sólo seis fueron sometidos a una reexposición intencionada al medicamento, observándose reaparición de las lesiones en cinco de ellos, y ausencia de ellas en un caso. Sólo dos de los 52 pacientes fueron sometidos al protocolo Naranjo,12 protocolo internacionalmente aceptado para evaluar la causalidad de las reacciones adversas a medicamentos basado en una escala de 10 preguntas relacionadas con la reacción adversa (tiempo de aparición, patrón de respuesta, retiro del medicamento, reexposición, etc.), obteniéndose un puntaje total y clasificándose en causalidad "definitiva", "probable", "posible" o "dudosa"; de los dos casos estudiados, la causalidad se clasificó como "posible" en el primero, y "posible o probable" en el segundo.11 Por lo anterior, es difícil establecer una conclusión definitiva y certera respecto a la relación medicamentosa asociada a esta enfermedad, ya que hacen falta información, algoritmos y estudios multicéntricos estandarizados que evalúen con más precisión esta causalidad.11
La lesión primaria de la enfermedad consiste en ampollas redondas, de contenido sérico o hemorrágico sobre piel normal, eritematosa, o placas urticariformes. Las ampollas son tensas, de tamaño variable y tienden a acomodarse en agrupaciones con un patrón "arciforme" denominado en "rosetas", "grupo de joyas" o "collar de perlas".2,4,10 El cuadro clínico es muy variado, inicialmente puede haber prurito y después aparecen las lesiones típicas, cuya localización más frecuente es en tórax y extremidades, lo cual también ocurrió en nuestro caso. Se ha señalado que también puede haber afectación de las mucosas causando ronquera, alteraciones faríngeas, y en casos excepcionales alteraciones oculares, queratoconjuntivitis y ceguera.3,4 Esta entidad patológica tiene una gran variabilidad clínica y se requiere de un estudio completo para poder realizar un buen diagnóstico y ofrecer el mejor tratamiento al paciente. La fisiopatología es aún muy incierta. Los aspectos inmunológicos de este padecimiento comprenden respuestas humorales y celulares. La respuesta humoral está determinada por la producción de autoanticuerpos IgA contra antígenos blanco en la membrana basal epidérmica depositados en disposición lineal. La mayoría de los pacientes desarrolla anticuerpos IgA contra los antígenos de 97 kD y de 120 kD, que son resultado de la proteólisis del antígeno BP180 del penfigoide ampolloso. El antígeno de 97 kD representa una porción del dominio extracelular del BP180, que se localiza en la lámina lúcida, mientras que el de 120 kD es una proteína en la membrana basal que se cree es producida por queratinocitos y funciona como proteína de anclaje.13-17 Además, se ha encontrado una molécula de 285 kD, que es un antígeno único y predominante en esta enfermedad y que difiere de la proteína de 290 kD, que corresponde al colágeno VII de las fibras de anclaje presente en la epidermólisis bullosa adquirida (EBA).1 Estudios más recientes por Talour y colaboradores han establecido una relación nueva (aunque muy poco común), con una proteína epidérmica de 200 kD.18 Por otra parte, la respuesta inmune celular está dada por los linfocitos T CD4, que al ser activados expresan marcadores de superficie como moléculas HLA-DR clase 1, moléculas de adhesión intracelular y vascular, e interleucinas.1 Su activación promueve el reclutamiento de neutrófilos y eosinófilos, que a su vez liberan citocinas que dañan la membrana basal. Los linfocitos Th2 contribuyen a la producción de citocinas proinflamatorias como IL4, IL5, IL8, que tienen efectos quimiotácticos de neutrófilos y eosinófilos.1,8 Como parte de la respuesta celular, el modelo más aceptado hasta hoy consiste en la activación del plasminógeno por los queratinocitos, y la activación de la metaloproteinasa 9 (MMP9) por los neutrófilos. La activación del plasminógeno, por su parte, contribuye a la formación de plasmina, que degrada el colágeno XVII para producir el antígeno de 97 kD antes mencionado, mientras que la activación de la MMP9 facilita que la elastasa de neutrófilos pueda inducir el desprendimiento dermoepidérmico para la formación de la ampolla.1
El diagnóstico de enfermedad por IgA lineal se sustenta en los hallazgos histopatológicos de la biopsia de piel perilesional, pero se confirma únicamente por la inmunofluorescencia directa, siendo ésta el estándar de oro.4 En la histopatología, pueden observarse ampollas subepidérmicas e infiltrado inflamatorio variable. No se sabe si la célula predominante del infiltrado inflamatorio depende del tiempo de evolución; sin embargo, se ha observado que los eosinófilos predominan en casos inducidos por medicamentos. En el estudio de inmunofluorescencia directa puede observarse un patrón linear y homogéneo de depósitos de IgA en la membrana basal de la piel perilesional. Este es el único inmunorreactivo en el 80% de casos, sin embargo, en el 20% restante pueden encontrarse también depósitos de IgG, IgM o C3 en la membrana basal.9 Además de los anticuerpos situados en la membrana basal, puede encontrarse una inmunofluorescencia indirecta positiva en el suero de hasta el 50% a 70% de los pacientes.18 Excepcionalmente también puede haber anticuerpos IgG circulantes.9,10 Algunos casos de enfermedad por IgA lineal pueden simular una dermatitis herpetiforme, sobre todo aquellos con presencia de microabscesos en la dermis papilar o con infiltrado de predominio de neutrófilos,4 la diferencia radica en que en la dermatitis herpetiforme se presenta una reacción inmunológica contra la gliadina (proteína estructural del gluten encontrada en trigo, centeno y cebada) y pueden hallarse anticuerpos circulantes IgA antitransglutaminasa tisular (TGt) y antiendomisio, siendo estos últimos los de mayor importancia para confirmar el diagnóstico.19 Al observar la inmunofluorescencia directa, los depósitos de IgA en la dermatitis herpetiforme son de forma granular en la punta de las papilas, mientras que en la dermatosis IgA lineal son lineales y homogéneos en la zona de la membrana basal.9 Otros casos de IgA lineal pueden simular un penfigoide ampolloso, sobre todo aquellos con predominio de eosinófilos en el infiltrado; sin embargo, en el caso de penfigoide ampolloso la inmunofluorescencia directa reporta principalmente depósitos de IgG y C3, y de IgA sólo en un 20%. Es importante descartar esta entidad patológica, para lo cual se deben incluir anticuerpos contra BPAg1 (230 kD) y/o contra BPAg2 (180 kD), por ELISA.4
La mayoría de los casos de enfermedad por IgA lineal responden muy bien al tratamiento con prednisona, dapsona o sulfapiridina.1 La respuesta comienza a verse en 48 a 72 horas, aproximadamente. Puede utilizarse dapsona como único tratamiento a dosis de hasta 300 mg/ día o sulfapiridina 1-1.5 g/día; también pueden emplearse dosis más bajas de medicamentos si se combinan con 5-30 mg de prednisona al día. Otra opción es sólo prednisona 15-30 mg al día. Para los pacientes pediátricos, se recomienda una dosis de sulfona de 1-2 mg/Kg/día o sulfapiridina 60/150 mg/Kg/día. Existen otros medicamentos que se han reportado como efectivos en caso de que no exista buena respuesta con los de primera elección, tales como colchicina, nicotinamida, tetraciclinas, inmunoglobulinas y doxiciclina.1,2 El interferón alfa ha sido utilizado exitosamente en casos severos,5 y recientemente se ha reportado la eficacia del uso de inmunoglobulina G intravenosa en dosis de 2-4 g/Kg por ciclo, cada cuatro semanas, combinado con dapsona y corticosteroides sistémicos, con una buena respuesta.20 El porcentaje de remisión va de un 10% a 60%, manifestado por muchos años de enfermedad, seguidos de una remisión espontánea.5,20
Conclusión
La dermatosis por IgA lineal es una enfermedad vesículo-ampollosa autoinmune poco común, en su mayoría idiopática, relacionada con enfermedades sistémicas o posiblemente asociada al uso de ciertos fármacos, aunque recientemente algunos autores han cuestionado esta última relación, debido a la falta de información y algoritmos adecuados de evaluación. Se han descubierto múltiples antígenos causantes de la enfermedad, sin embargo los más estudiados son una proteína de 97 kD en la lámina lúcida y una proteína de 120 kD, ambas son producto de la degradación del antígeno del penfigoide ampolloso BP180, de ahí la similitud de las manifestaciones cutáneas. Dada la heterogeneidad de la enfermedad, deben realizarse múltiples estudios para poder establecer el diagnóstico, el cual se confirma por la presencia de anticuerpos de IgA lineales en la membrana basal en la inmunofluorescencia directa. El tratamiento de elección son las sulfonas y éste tiende a ser muy satisfactorio, mostrando resultados desde la primera semana. En el caso expuesto anteriormente, la paciente mostró mejoría parcial de las lesiones previas y ausencia de nuevas lesiones tras una semana de tratamiento con medicamentos tópicos más sulfona a dosis leve de 100 mg al día, aumentando la eficacia tras el reajuste de dosis con 200 mg al día y logrando una remisión completa después de algunos meses de tratamiento. Se debe tomar en cuenta que esta enfermedad puede simular otros desórdenes ampollosos, y que si bien el tratamiento en todos ellos sería similar, la etiología y las manifestaciones clínicas son distintas, por lo cual es indispensable realizar un buen diagnóstico basado en el interrogatorio, la clínica y los estudios pertinentes de laboratorio y gabinete.
Conflicto de intereses
Los autores declaran no tener conflicto de intereses.
Financiamiento
Los autores no recibieron ningún patrocinio para llevar a cabo este artículo.
Agradecimientos
Al Lic. David Ahedo por su asistencia en la búsqueda de información vía electrónica.
Recibido: Septiembre 2012.
Aceptado: Julio 2013
Correspondencia:
Julio C. Salas Alanís.
Servicio de Dermatología, Hospital Universitario "Dr. José Eleuterio González", Universidad Autónoma de Nuevo León.
Otomí N° 206, Colonia Azteca, Guadalupe, C.P. 67150, Monterrey, N. L., México.
Teléfonos: (+52) 81 8367 6060 y (+52) 81 8253 5943.
Correo electrónico: drjuliosalas@gmail.com