


Escolar femenina de seis años de edad, quien inicia en febrero de 2011 con crisis convulsivas motoras tónico-clónico generalizadas, presenta un electroencefalograma con actividad irritativa focalizando inicialmente en hemisferio izquierdo con rápida generalización y sincronización secundaria, la cual es continua, de predominio en sueño sin movimientos oculares rápidos (NMOR), con inadecuado control de sus crisis en tratamiento con fenitoína y períodos de ausencia de breve duración, que aumentan en frecuencia, motivo por el cual acude a nuestro hospital. Se cambia el medicamento a valproato de magnesio con una mejor respuesta. Su exploración neurológica interíctica es normal y la imagen por resonancia magnética de cerebro sin alteraciones estructurales. Se hace el diagnóstico de epilepsia criptogénica y electroencefalograma compatible con estado epiléptico eléctrico del sueño (ESES, por sus siglas en inglés). En conclusión, se considera el ESES como una condición electroencefalográfica que debe ser estrechamente correlacionada a la clínica para su diagnóstico.
A 6-year-old female starts in February 2011 with generalized tonic-clonic seizures. The EEG showed irritative activity focusing initially on the left hemisphere with rapid secondary generalization and synchronization which is predominantly continuous in NREM (non rapid eye movement sleep), with inadequate control of seizures with monotherapy (phenytoin), and with an increase in the frequency of absence episodes, reason for why she comes to our hospital. Therapy was changed to magnesium valproate with better response; her interictal neurological examination was normal, brain MRI without structural abnormalities, negative neurological signs not present. A diagnosis of cryptogenic epilepsy was done and an ESES diagnosis was made by EEG. In conclusion, ESES is an electroencephalographic condition that must be closely correlated to the clinic for its diagnosis.
Introducción
Estado Epiléptico Eléctrico en Sueño (ESES) es una forma especial del estado epiléptico que representa actividad irritativa comicial en el electroencefalograma, manifestado en etapas de sueño pasivo (NMOR) del trazado, dicha actividad es suprimida en estados de vigilia. Al valorar un paciente con crisis comiciales en presencia de esta actividad eléctrica, es necesario realizar una historia clínica completa, clasificar la semiología de las crisis convulsivas y valorar los hitos del desarrollo alcanzados para su edad, así como un posible síndrome de regresión o alteraciones del lenguaje concomitantes, con motivo de clasificarlo como un síndrome epiléptico y con ello poder facilitar el diagnóstico definitivo, así como la respuesta al tratamiento y el pronóstico que podemos ofrecer al paciente.
El caso que presentamos muestra este patrón electroencefalográfico de actividad epiléptica en sueño, con los datos negativos relevantes de alteraciones del lenguaje (principalmente afasia de predominio sensitivo), que hace diagnóstico de síndrome de Landau Kleffner, llamado también afasia epiléptica adquirida. También pueden presentarse datos de síndrome de regresión (demencia infantil), que a su vez integra el síndrome conocido con el acrónimo de CSWS (por sus siglas en inglés, Continuous Spike Wave during Sleep) o actividad continua de punta onda lenta, durante el sueño NMOR. En conjunto, estas dos entidades dentro de los hospitales de tercer nivel o centros de referencia de epilepsia es bajo, comprendiendo desde el 0.3% a 2% de todas las epilepsias. Esta actividad registrada en el electroencefalograma, sin los datos clínicos mencionados hace relevante el caso clínico de nuestra paciente.
Presentación del caso
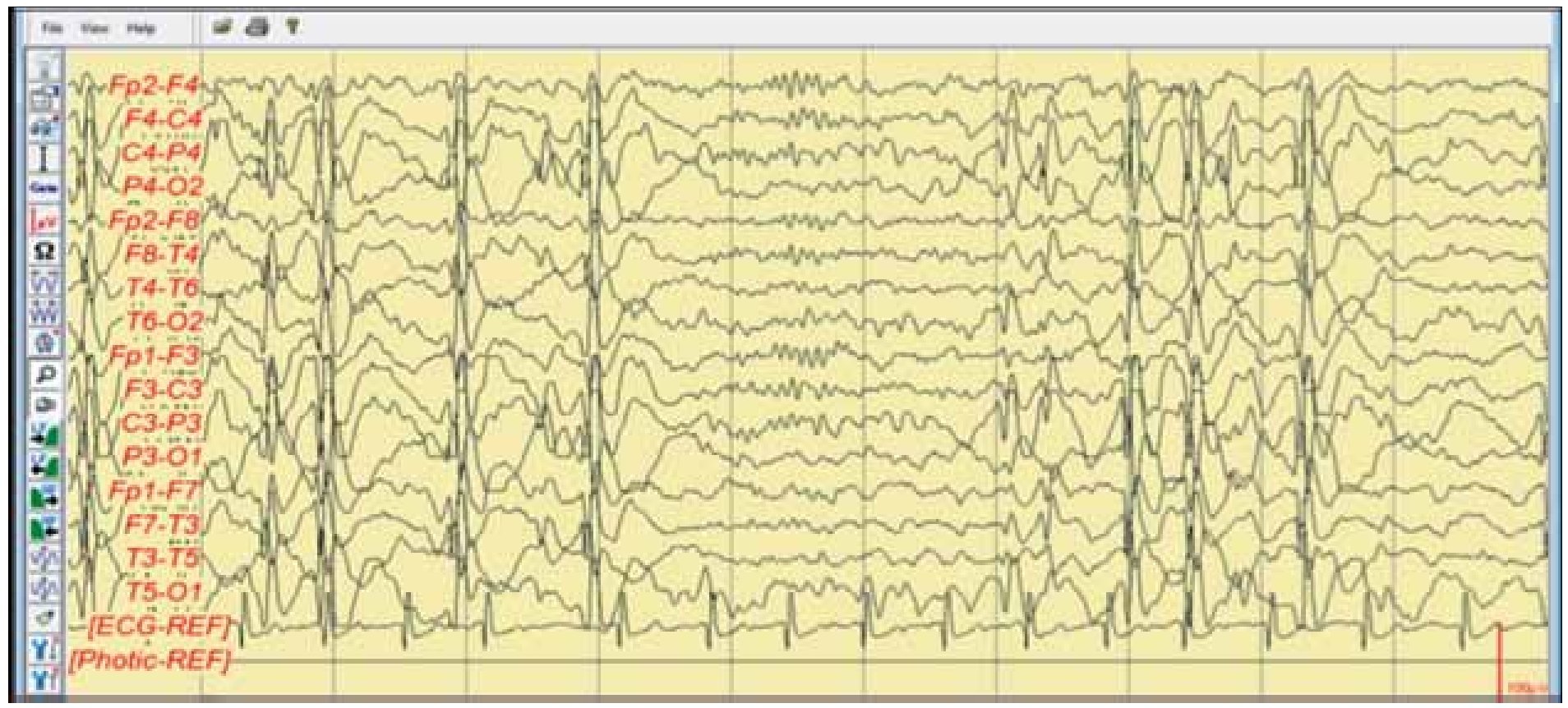
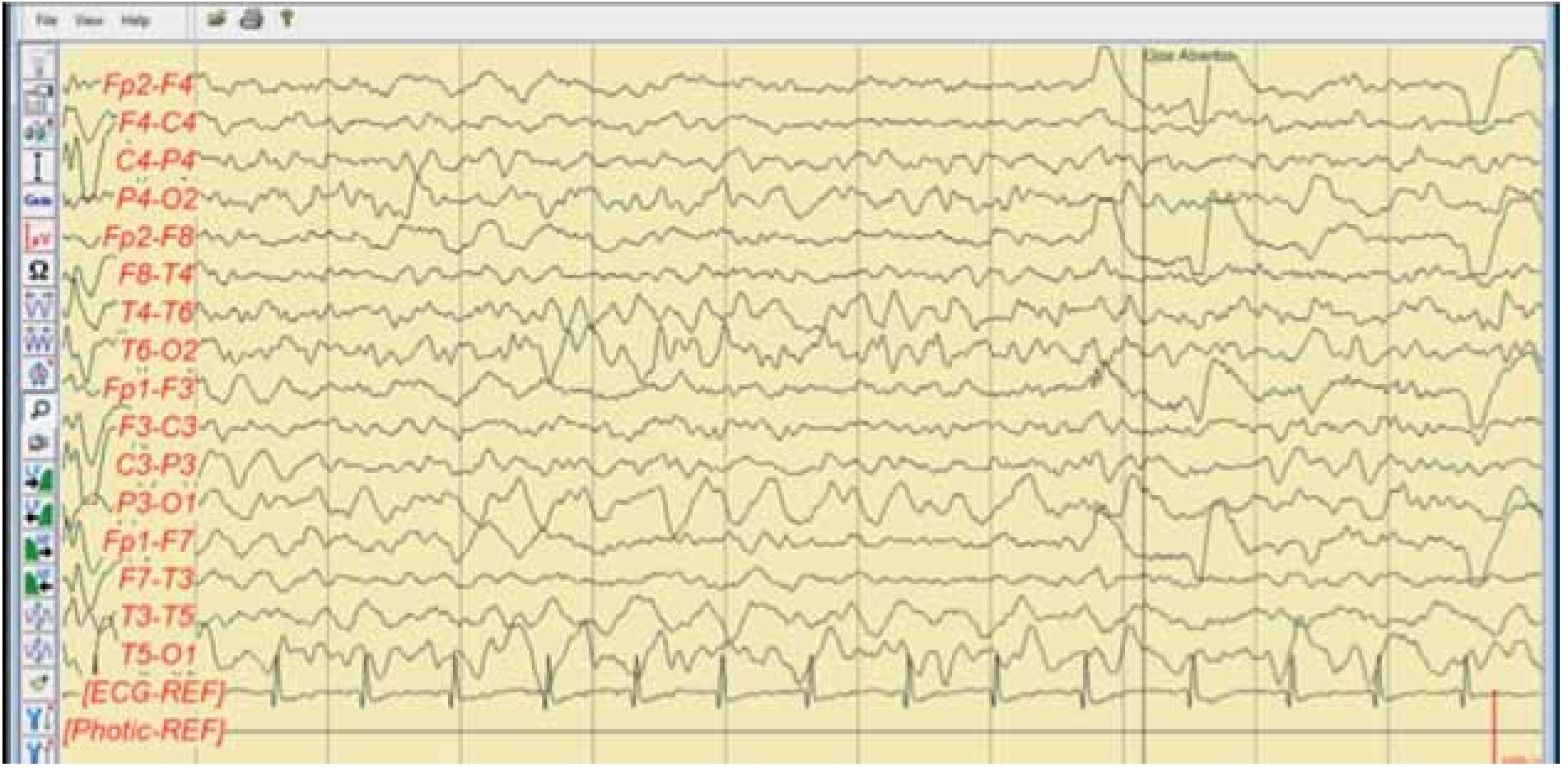
Paciente femenina de seis años de edad, con antecedentes heredofamiliares de un primo de 15 años, rama materna, con epilepsia en tratamiento, antecedentes perinatales y personales patológicos negativos, con un neurodesarrollo normal para la edad caracterizado por presentar lenguaje en forma y función (fonología, léxico, sintaxis, pragmática y cognitiva) sin alteraciones, lateralidad diestra, aprovechamiento escolar en la educación preprimaria sobresaliente, juego imaginativo y en conjunto, adecuado por apreciación del maestro del kínder. En febrero de 2011, el paciente inicia por la mañana con un episodio de crisis convulsiva caracterizada por movimientos de tipo clónico de miembro superior derecho, sin alteración del estado de alerta, con una generalización secundaria tipo tónico-clónica, con duración aproximada a cinco minutos, periodo postictal prolongado, somnolencia de tres horas y recuperación íntegra, se evalúa en un servicio de urgencias iniciando tratamiento con fenitoína a dosis de 5 mg/kg/día, se solicita electroencefalograma para su estudio. El electroencefalograma muestra una actividad con ritmo de fondo maduracional con ojos cerrados en vigilia, adecuado para la edad de la paciente y en sueño, llama la atención la presencia de actividad comicial tipo punta onda lenta de 1 a 3 Hz bilateral sincrónica y simétrica, que continúa en la mayor parte del trazado electroencefalográfico, de topografía predominante hacia cuadrantes posteriores o regiones occipitales, con mínimo involucro hacia canales frontales bilaterales (Figura 1), que se suprime al encontrarse en vigilia (Figura 2). Ya con tratamiento clínicamente, la madre observa la presencia de alteraciones del estado de alerta (refiriéndolas como muy distraída), caracterizadas por falta de respuesta a estímulos verbales de menos de 30 segundos de duración, que en ocasiones se acompañaron de automatismos faciales en estado de vigilia.
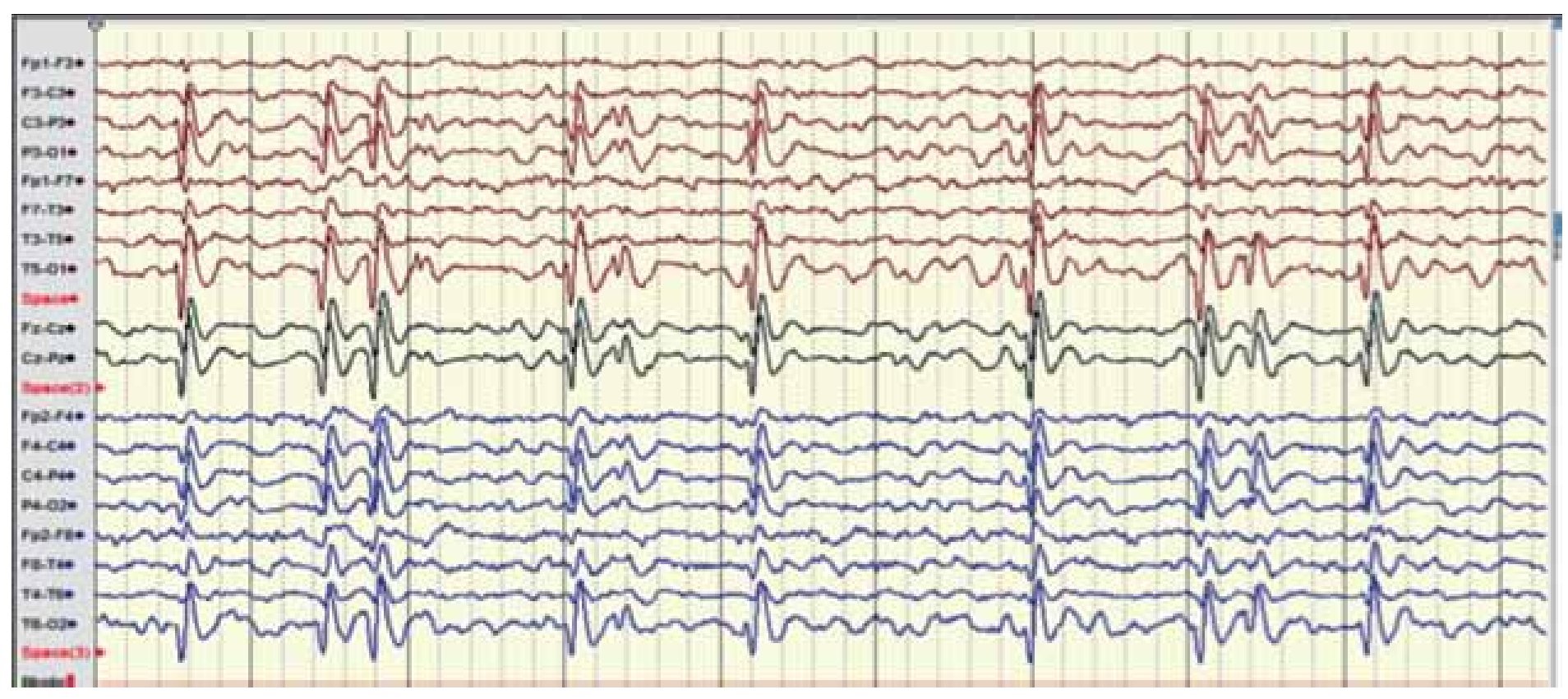
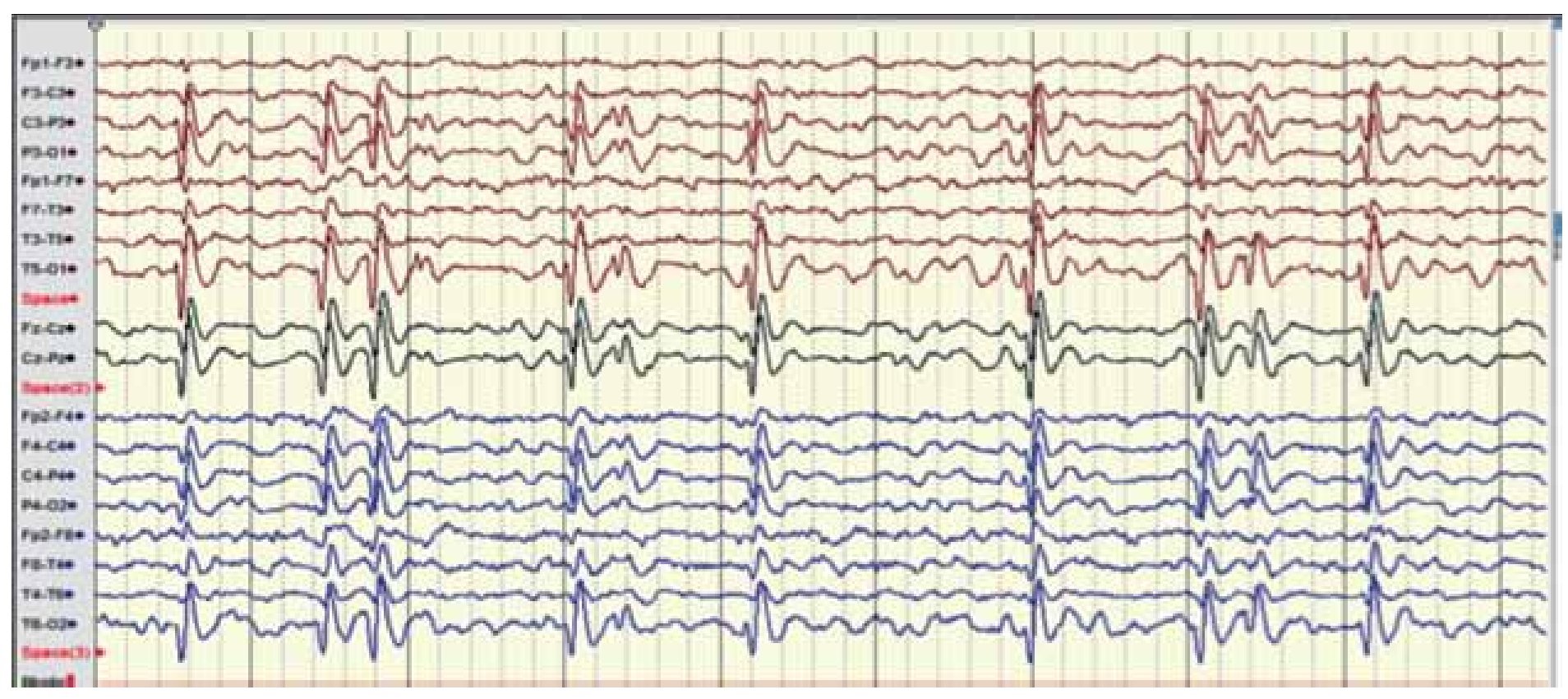
Figura 1. Electroencefalograma digital realizado en marzo 2011. EEG digital con montaje bipolar y longitudinal, donde se aprecia un ritmo de fondo hacia regiones posteriores en rango tetha-delta compatible con sueño NMOR, y presencia de actividad irritativa bilateral y sincrónica tipo punta, onda lenta variable en frecuencia de 1-2 Hz con mayor topografía hacia cuadrantes posteriores, con mínimo involucro de canales frontales bilaterales.
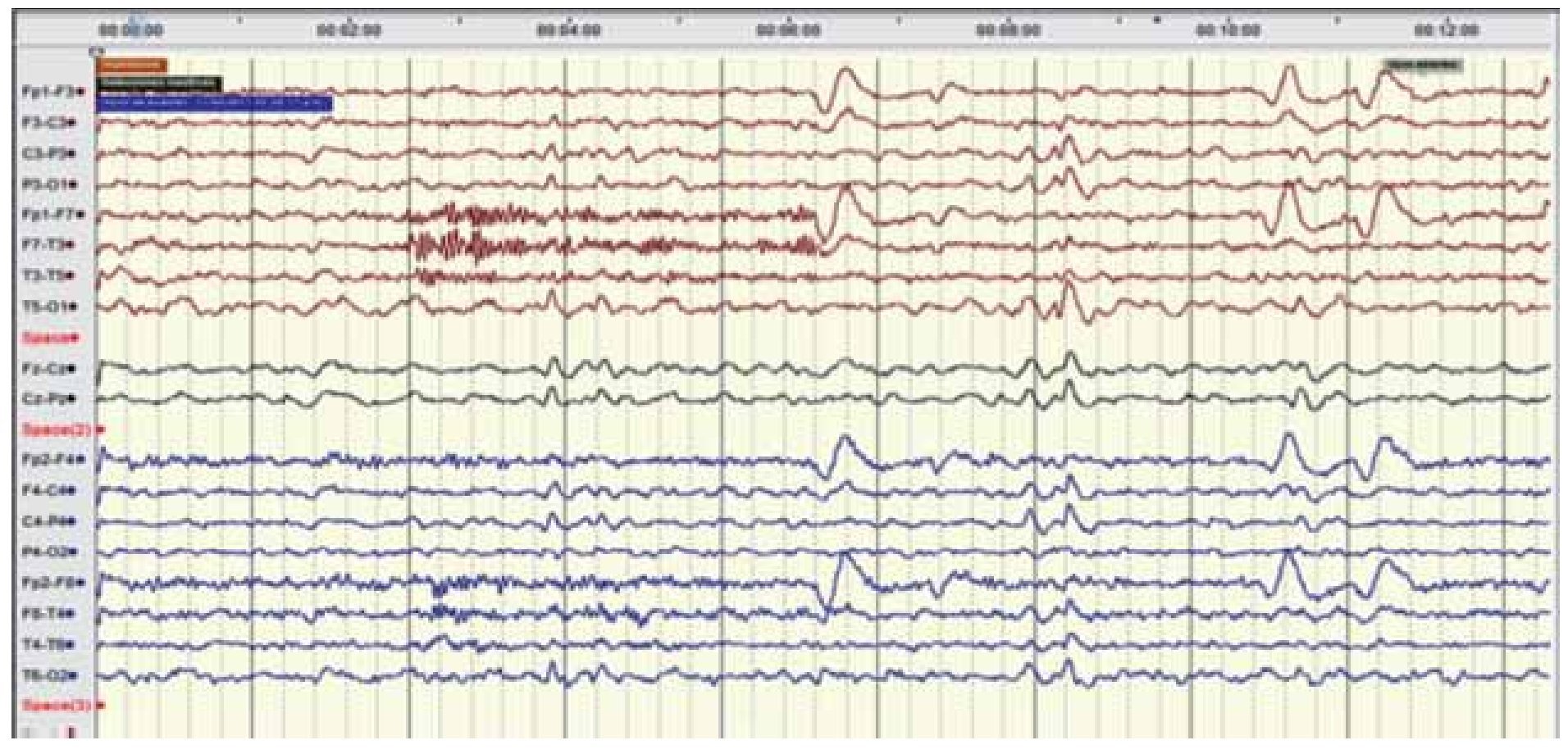
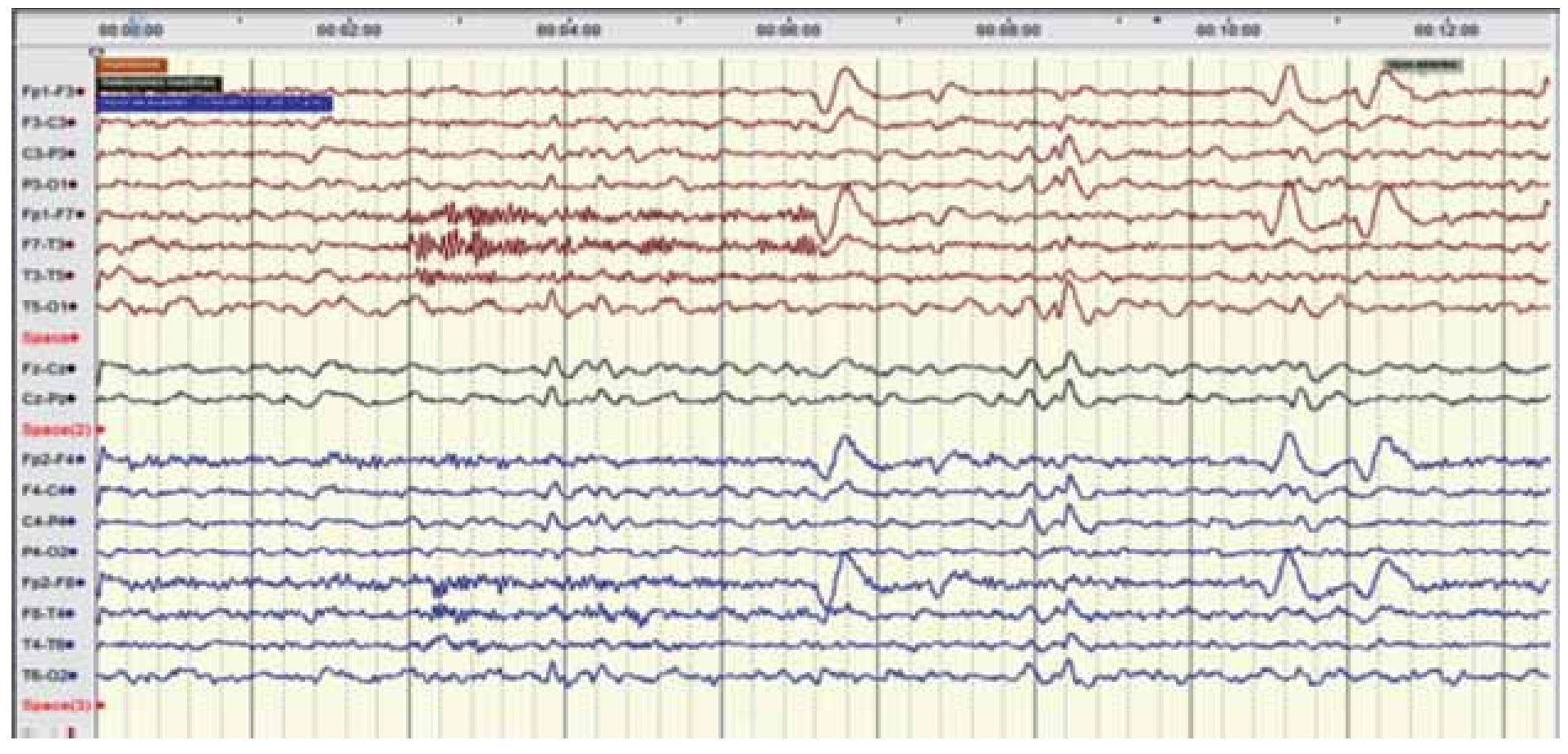
Figura 2. EEG digital marzo 2011. Trazado longitudinal bipolar que permite ver la ausencia de actividad convulsiva en estado de vigilia, observando adecuado gradiente anteroposterior con actividad miogénica en canales frontales bilaterales en el segundo 4, y artefacto de parpadeo en canales frontales en el segundo 6 y 10.
Durante el periodo de cuatro meses, presenta tres episodios de eventos tónico-clónico generalizados, de las mismas características ya descritas (de inicio focales en hemicuerpo derecho secundariamente generalizadas), además de referir que en sueño observan movimientos ocasionales de tipo clónico, siempre de hemicuerpo derecho, de breve duración. Su rendimiento escolar y sus capacidades motoras finas, gruesas y la esfera social se mantuvieron preservadas durante este lapso. Se mantiene con el tratamiento ya iniciado. Acuden al servicio de neurología pediátrica para una segunda opinión, con un nuevo trazado electroencefalográfico analógico, en esta ocasión muestra descargas que predominan en la mayor parte del trazado de la misma topografía y en sueño (Figura 3), una imagen por resonancia magnética cerebral sin alteraciones estructurales (Figuras 3 a 5). Se decide suspender gradualmente fenitoína la cual se encontraba en niveles séricos subterapéuticos 9 mcg/ml (valores normales 10-20 mcg/ml) e iniciamos valproato, a dosis inicial de 15 mg/Kg/día, dos veces al día. Cinco días posteriores acuden nuevamente al servicio de Urgencias pediátricas de nuestro Hospital, al presentar un evento de alteración del estado de alerta, sin asociarse a proceso infeccioso o falta de administración del medicamento, con ausencia de respuesta a estímulos visuales, sonoros y táctiles mayor a 20 minutos de duración. A su arribo con signos vitales normales sin postura característica, no había lesiones en piel ni datos infecciosos en oídos o faringe, sistema cardiopulmonar sin compromiso, sin reactividad al dolor con la mirada conjugada central, y que en ocasiones se desviaba de manera clónica hacia la derecha, pupilas isocóricas normoreactivas a la luz, fondo de ojo normal, sin asimetría facial, sensibilidad, coordinación, habla, marcha y fuerza no valorables, reflejos de estiramiento con hiperreflexia generalizada sin clonus, respuesta plantar indiferente, sin signos meníngeos. Con esta descripción, se diagnostica estado epiléptico parcial complejo.
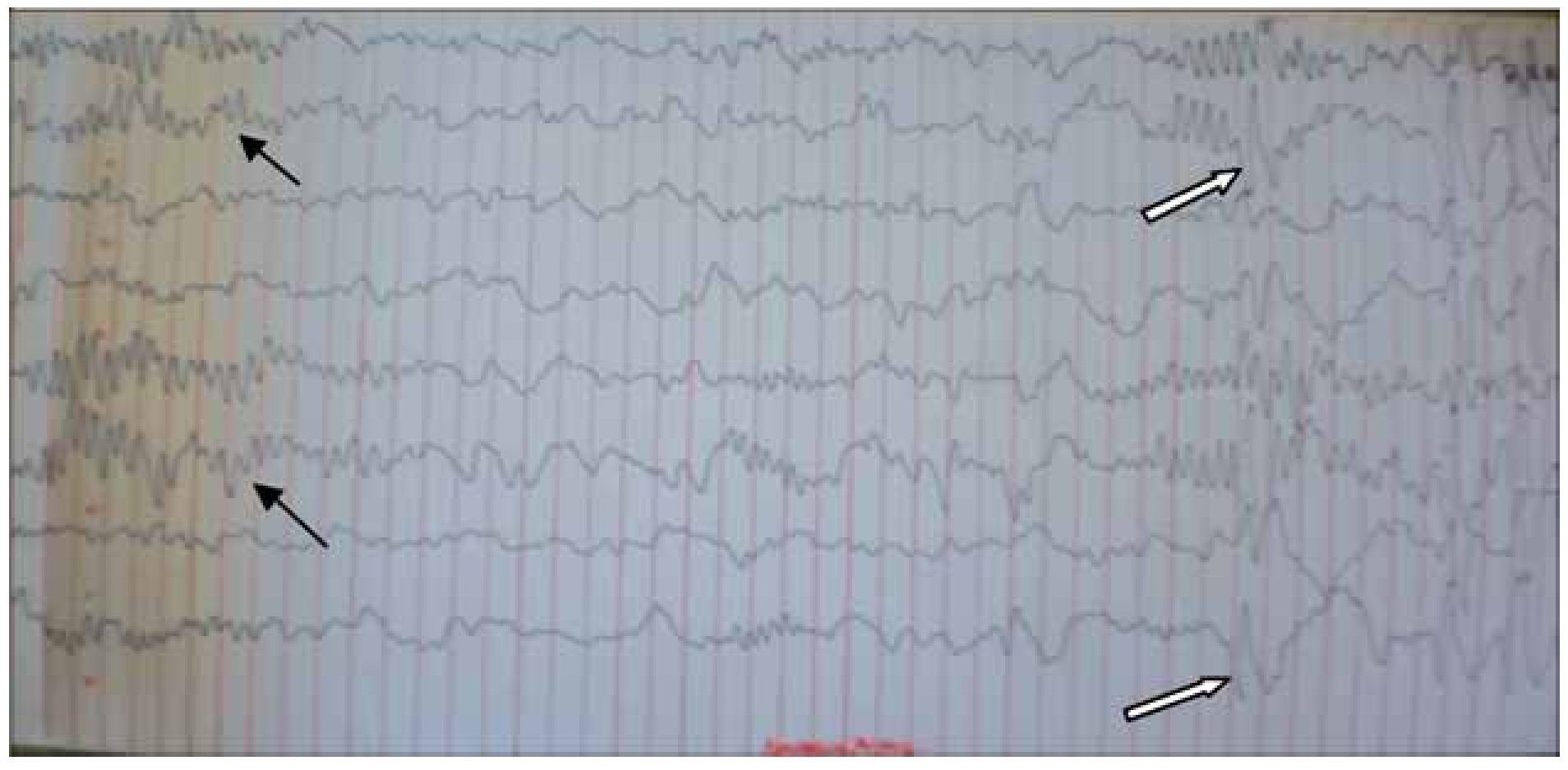
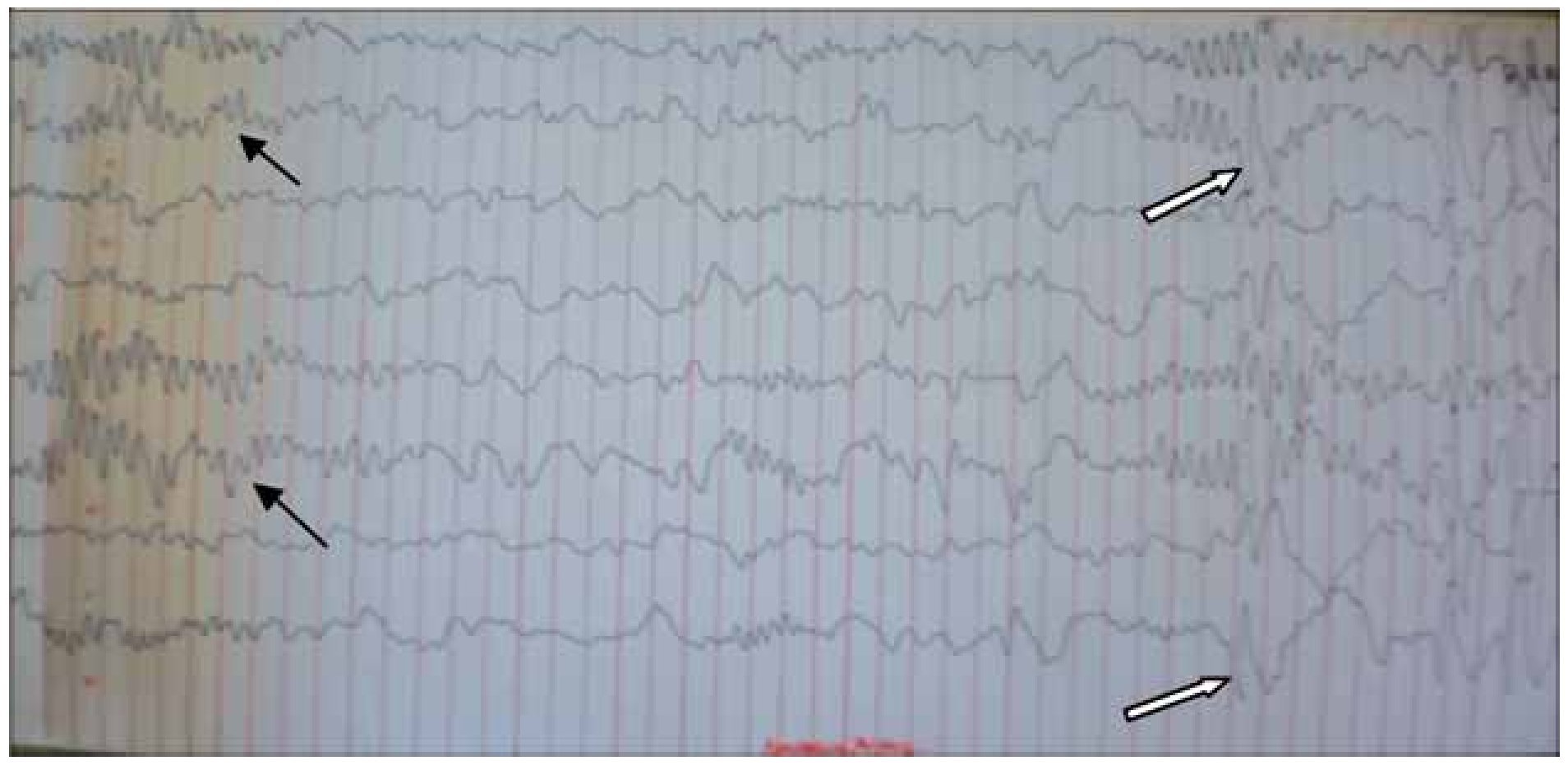
Figura 3. Electroencefalograma analógico junio 2011. EEG analógico en montaje bipolar parasagital, primeros cuatro canales de arriba hacia abajo derechos y siguientes 4 izquierdos. Se aprecia un ritmo de fondo lento en regiones posteriores en rango tetha-delta, hacia cuadrantes anteriores la formación de husos de sueño (flechas negras), de manera sincrónica y simétrica que nos ubican en sueño etapa 2, casi al final del trazado nuevamente la actividad irritativa punta onda lenta (flechas blancas).
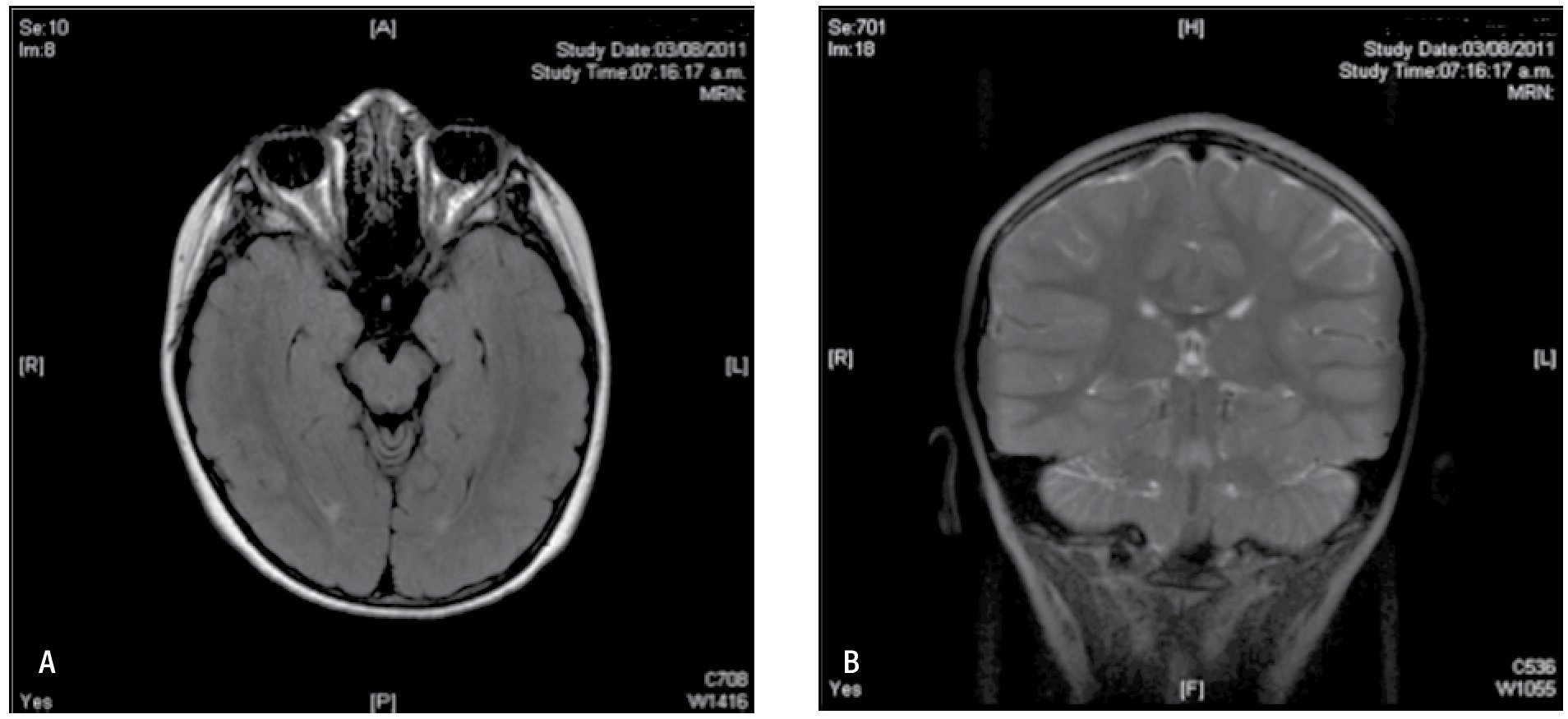
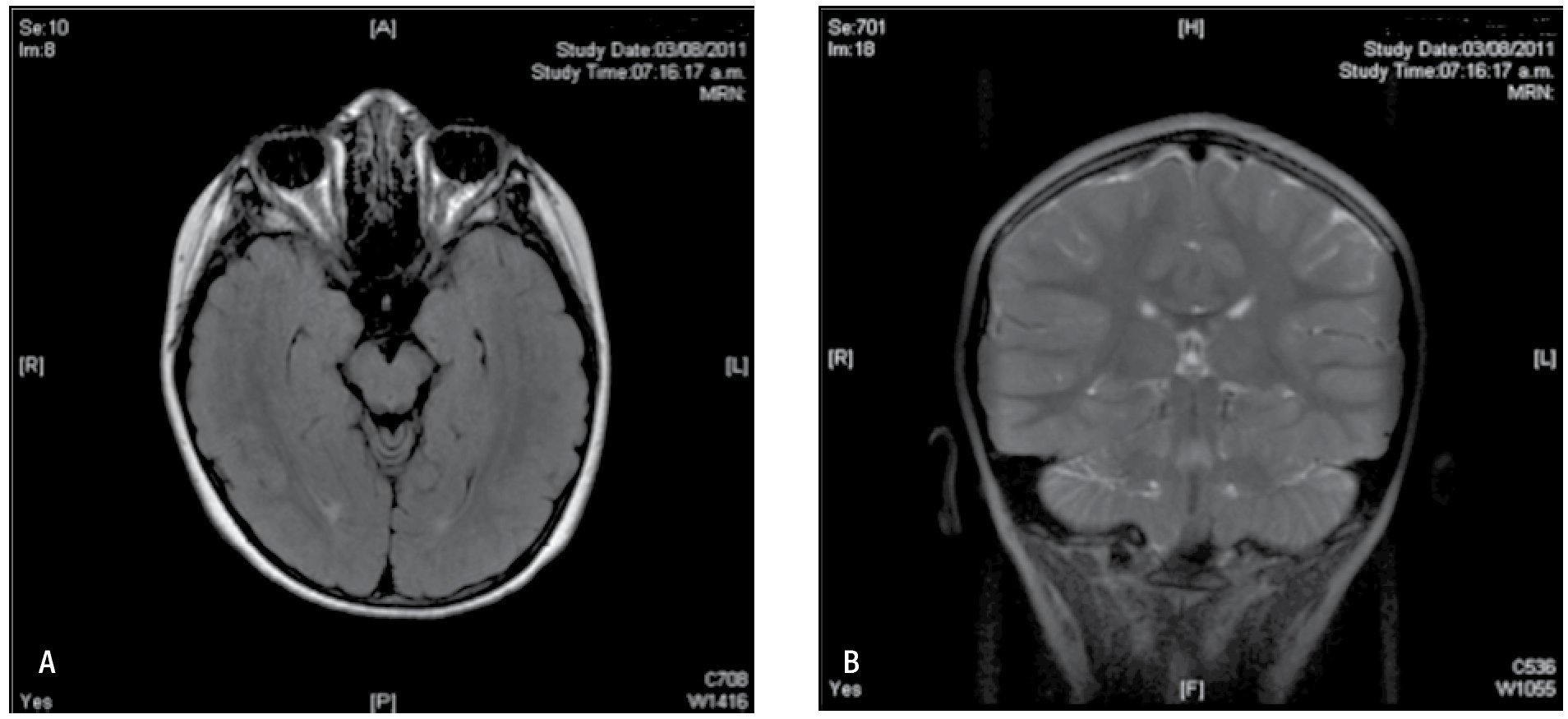
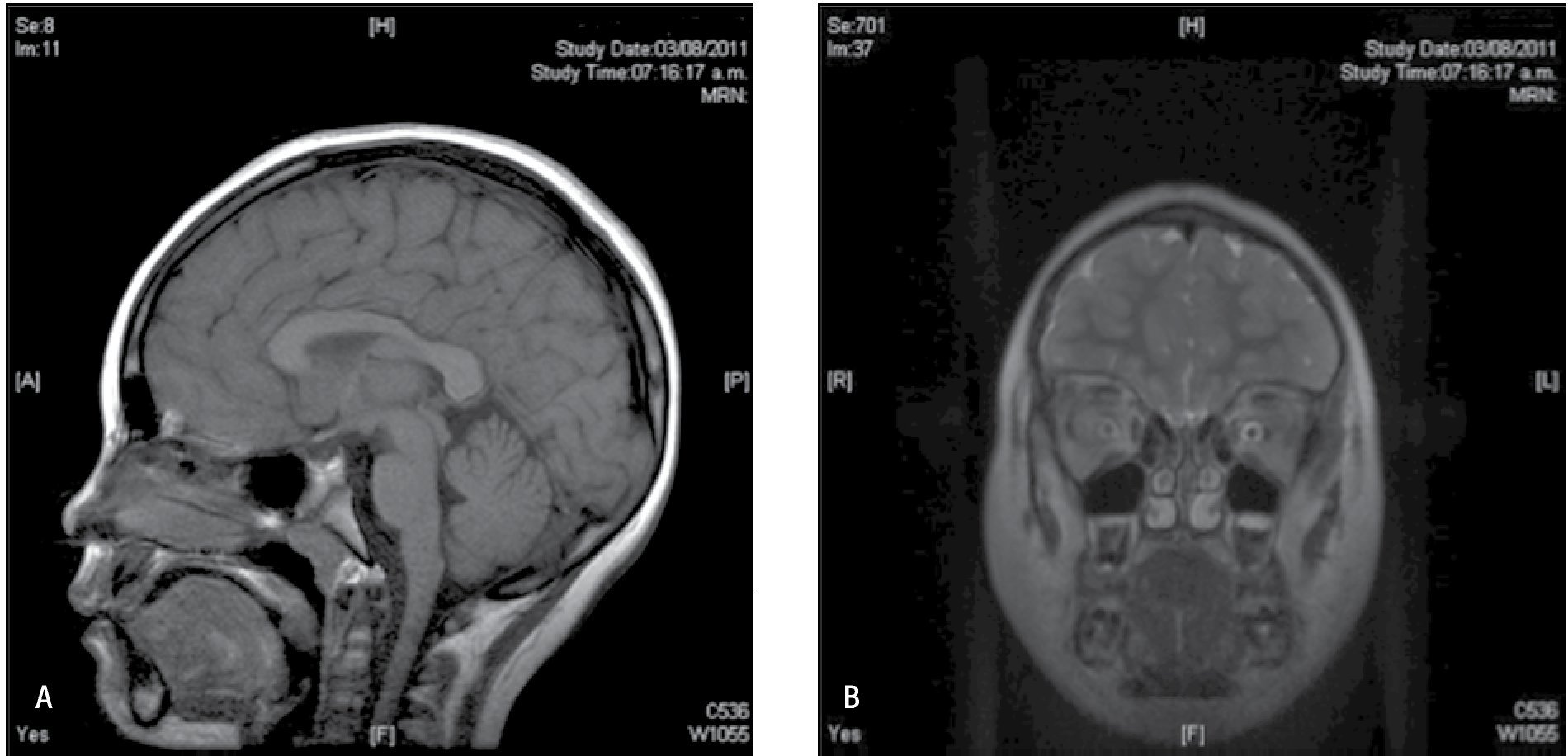
Figura 4. IRM de cerebro agosto 2011. A) IRM de cerebro simple en corte axial con secuencia T1 que permite ver a lateral a ambos pedúnculos cerebrales ambos hipocampos simétricos sin alteraciones en su intensidad. B) Corte coronal de IRM de cerebro en secuencia T2 que también demuestra la integridad de la corteza mesial temporal, una asimetría o alteración en la intensidad permitiría orientarnos a una etiología sintomática en el padecimiento de nuestra paciente.
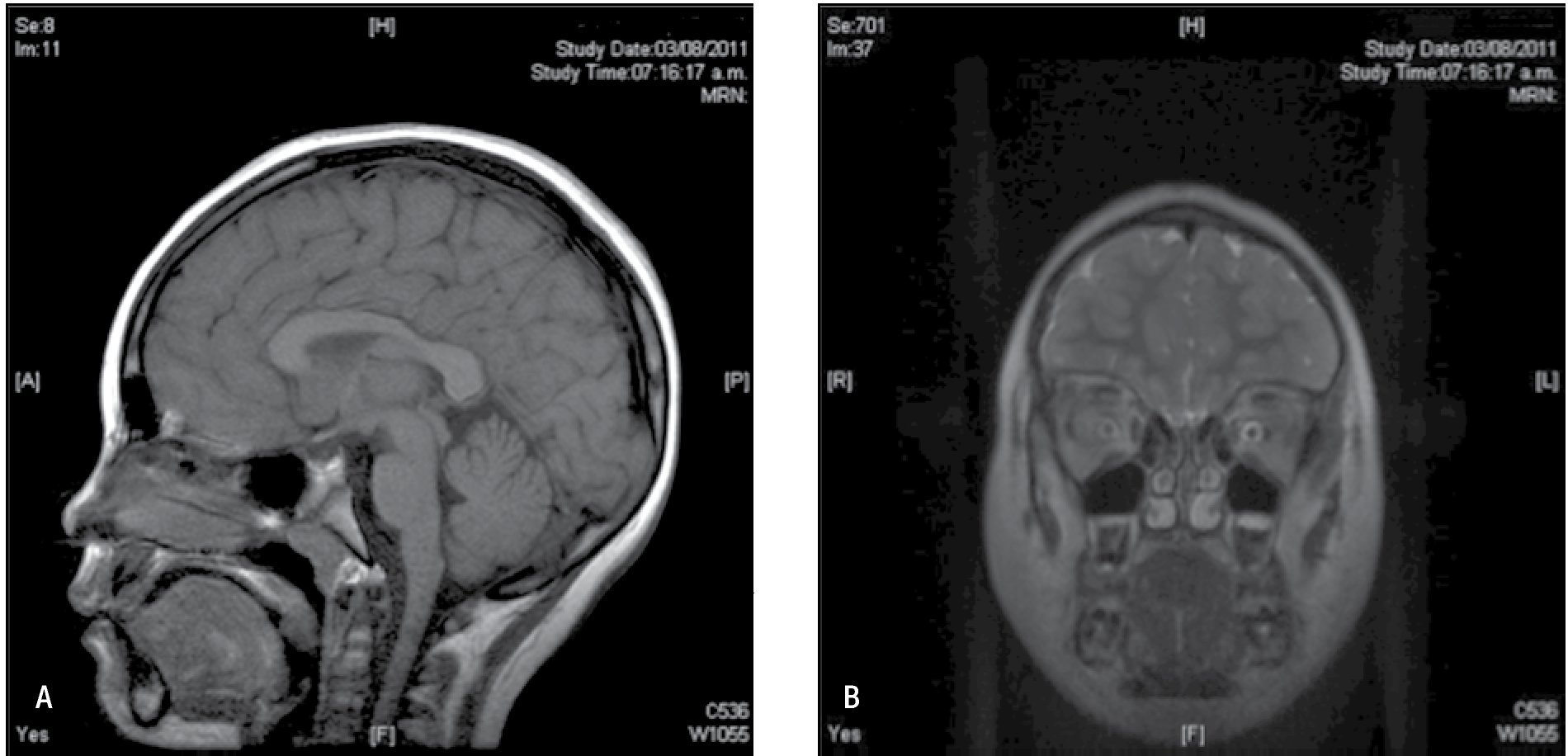
Figura 5. IRM de cerebro agosto 2011. A) IRM de cerebro simple corte sagital en la línea media con secuencia en T1, sin alteraciones estructurales. B) IRM de cerebro simple en T2 con corte coronal a nivel de región orbitaria, que permite valorar ambas cortezas frontales las cuales se encuentran íntegras en su anatomía.
Se yugula con monodosis de diacepam a 0.1 mg/Kg/ dosis sin complicación, con un postictal de somnolencia de dos horas y con recuperación íntegra, completando el resto de la exploración neurológica sin datos de focalización. Se solicitan estudios generales: biometría hemática con hemoglobina 12 g/dL, cuenta leucocitaria 7.000/mm3 70% PMN, plaquetas 230.000/mm3, pruebas de funcionamiento hepático AsT/ALT 28/42 y electrólitos séricos Na 135, K 4.2, Ca 8.9. Niveles séricos de valproato en rango infraterapéutico 33 mcg/mL (valores normales de 50-100 mcg/mL). Se suspende completamente la fenitoína, se impregna nuevamente con valproato 15 mg/Kg/dosis y se incrementa la dosis a 30 mg/Kg/día tres veces al día, ingresando a sala de internamiento para su observación. Se realiza video-EEG que presenta la actividad irritativa sin correlación clínica ya antes descrita, compatible con diagnóstico eléctrico de ESES (Figuras 6 y 7), tras dos días de observación sin nuevos eventos motores ni alteración del estado de alerta, se egresa en condiciones estables para su control en la consulta externa de neurología pediátrica, con monoterapia basada en ácido valproico a dosis de 40 mg/kg/día, con resultado de pruebas neuropsicológicas incluido coeficiente intelectual para continuar su evolución.
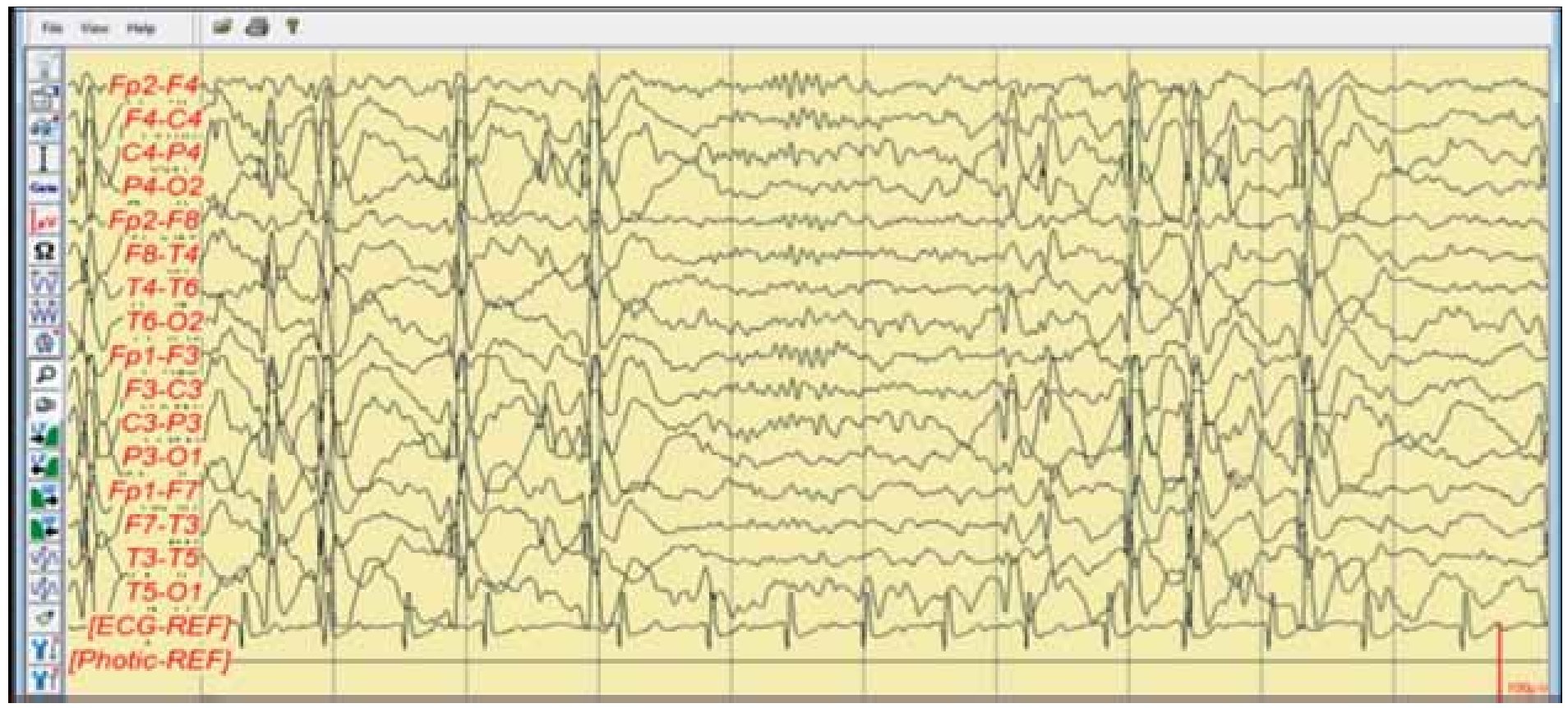
Figura 6. Electroencefalograma digital agosto 2011. Electroencefalograma que muestra el patrón ESES, justo en el centro del trazado se aprecian bien formados los husos de sueño, que nos ubican en la fase 2 de sueño y la actividad comicial presente en más del 80% de trazado.
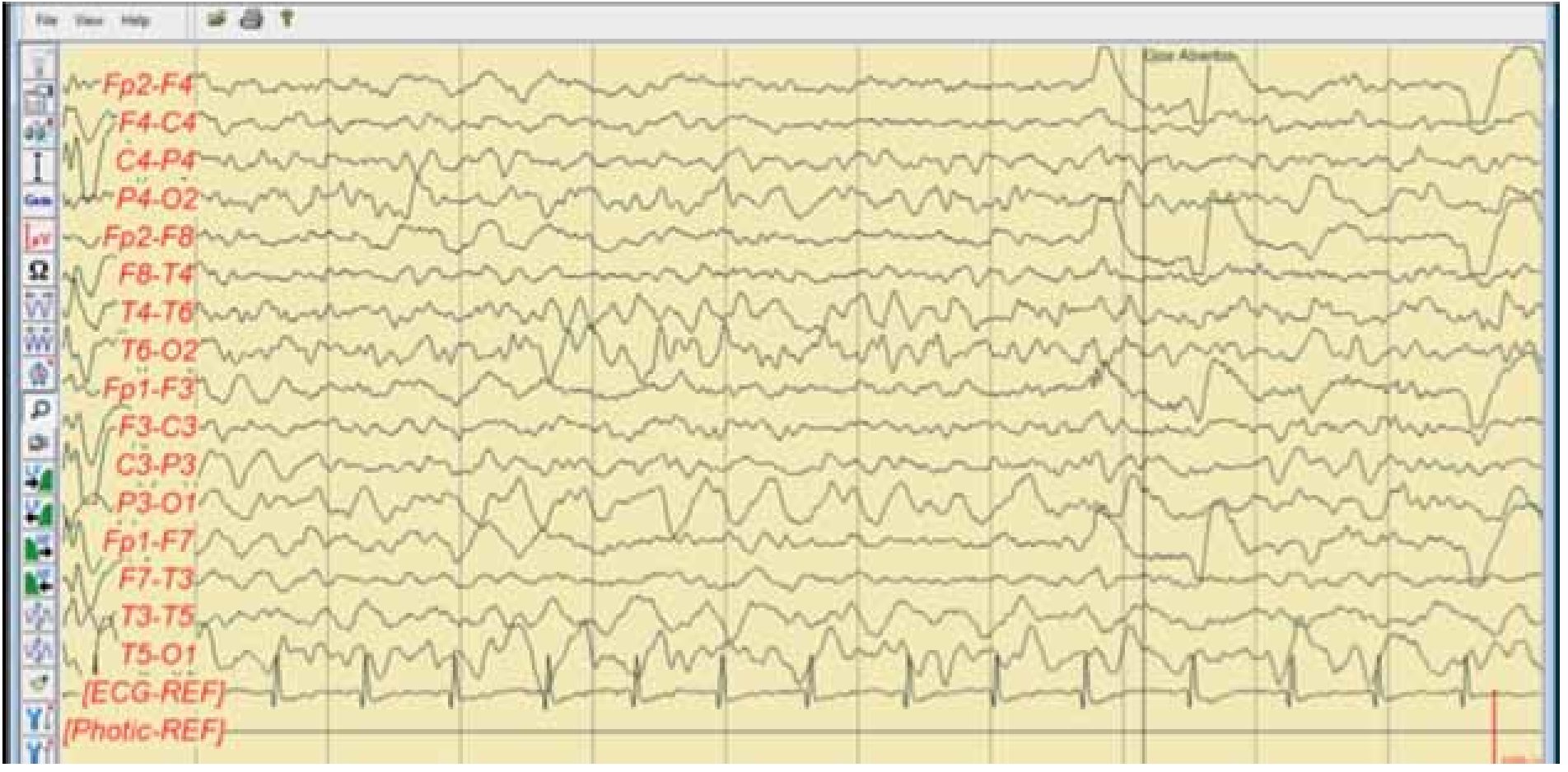
Figura 7. Electroencefalograma digital Hospital Universitario agosto 2011. Electroencefalograma en vigilia, se aprecia una asimetría interhemisférica con ritmos más lentos en rango delta, predominantes hacia hemisferio izquierdo, además que en vigilia no presenta la actividad comicial antes descrita. Esta actividad corresponde a la focalidad clínica convulsiva de su hemicuerpo derecho.
Se obtuvieron mediante sesiones semanales, los resultados de pruebas neuropsicológicas, realizando Escala Internacional de Inteligencia Infantil de Wechsler (WPPsI), Evaluación Neuropsicológica Infantil (ENI), Cuestionario Vanderbilt para TDAH y test de Bender.
Sus resultados para WPPsI fueron de 110 para escala verbal, escala de ejecución 88 y escala de inteligencia global 99 (todos dentro de límites normales), ENI en memoria, percepción táctil, lenguaje, escritura, y aritmética arriba del promedio para la edad, escala de edad cronológica y maduracional de Bender adecuada. Por último presentó criterios para TDAH por cuestionario de Vanderbilt de predominio inatento-hiperactivo, el cual fue el diagnóstico por parte de neuropsicología.
Discusión
De los síndromes epilépticos de la clasificación de la ILAE (por sus siglas en inglés, International League Against Epilepsy) englobados dentro de formas especiales del estado epiléptico, se encuentran el síndrome de Landau Kleffner y CSWS (Continuous Spike Wave in Sleep), que se caracterizan por presentar el trazado electroencefalográfico conocido como ESES,1 el criterio para considerarse el diagnóstico es por una actividad paroxística con puntas ondas lentas simétricas, sincrónicas generalizadas durante el sueño NMOR, que varían en su proporción del 25% al 85% del trazado,2,3 un dato adicional propuesto por Tassinari es que debe de tener al menos 3 electroencefalogramas, en un periodo de un mes con estas alteraciones. La conjunción de ESES más trastorno en el lenguaje específicamente afasia de tipo sensitivo o regresión del neurodesarrollo, hacen clasificar en uno de estos dos síndromes epilépticos (SLK o CSWS, respectivamente), que se consideran benignos en cuanto a su respuesta a fármacos antiepilépticos, no así sus posibles secuelas a nivel cognitivo.
En el caso presentado, clínicamente la edad escolar de la paciente corresponde al inicio de este tipo de padecimientos que hacen su debut de los cuatro a los 11 años, sin preferencia por un género sobre otro.2,4 El antecedente familiar del primo con epilepsia resalta sobre el resto que fue negativo al interrogatorio intencionado, niegan eventos de insulto en el periodo perinatal, lo que deja en duda una posible causa etiológica idiopática (recordando que sólo en epilepsia a diferencia del resto de las etiologías en medicina, el término idiopático se refiere a causas genéticas), la semiología de las crisis incluye varios tipos de crisis comiciales además de las tónico-clónico generalizadas, que son las causantes de las visitas al Servicio de Urgencias de los padres. Los episodios de alteración del estado de alerta de breve duración con automatismos faciales sin periodo postictal, que se exacerbaron con el primer tratamiento recetado (fenitoína) es compatible con crisis de ausencia que a posteriori con el electroencefalograma corresponden a ausencias atípicas, y por último, los eventos clónicos nocturnos de hemicuerpo derecho ocasionales, así todos estos eventos se evaluaron y concluimos que presenta múltiples tipos de crisis convulsivas, que hacen ubicar un posible foco epileptógeno hacia regiones prerrolándicas motoras en hemisferio izquierdo para las motoras focales secundariamente generalizadas, corteza motora suplementaria frontal izquierda por la desviación de la mirada hacia la derecha, o bien corteza frontal, núcleos amigdalinos del hipocampo o subcorticalmente en el sistema reticulocotical por las crisis de ausencia atípica,5 para su evaluación de una posible etiología estructural, la IRM no demostró alteraciones en intensidad en ninguna de estas regiones, su trazado en los tres electroencefalogramas realizados coinciden en la actividad de ESES al estar presente en casi 90% del trazado de sueño, misma actividad que no se observa en estado de vigilia con topografía predominantemente hacia los cuadrantes posteriores y mínimo involucro hacia regiones frontales bilaterales, dato eléctrico compatible más hacia los trazados descritos en los pacientes con síndrome de Landau-Kleffner,6-8 sin complementarnos su dato clínico pivote que es afasia sensitiva del lenguaje, el tratamiento se inicia con valproato escogido por su capacidad no sólo antiepiléptica sino por la capacidad anticonvulsivogénica,9 como monoterapia a dosis altas tratando de mantener los niveles séricos arriba de 90 mcg/mL, opciones de tratamiento son carbamacepina, benzodiacepinas como diacepam o clobazam tanto solos como biterapia, los fármacos anticomiciales de segunda generación probados son lamotrigina y levetiracetam.6 No hay un consenso uniforme sobre el tratamiento, pues ciertas series de casos inician con dosis altas de diacepam (1 mg/kg/dosis rectal o vía oral) durante un mes o incluso esteroide (ACTH si se cuenta con el medicamento), o bien prednisona iniciando a dosis altas con disminución gradual durante seis hasta 21 meses.10,11 De manera que el objetivo principal no es la disminución de las crisis, las cuales son de fácil control, sino la normalización del electroencefalograma, el cual puede ser tan rápido en semanas de tratamiento como tan tardío hasta cuatro años de haber iniciado el padecimiento o hasta la pubertad, un factor de buen pronóstico en la paciente es que su padecimiento inició después de la edad preescolar, pues está demostrado que un debut de síntomas de regresión en edades tempranas se relaciona a peor pronóstico funcional.12 Se considera que si el trazado de ESES persiste dos años después de haber iniciado tratamiento anticomicial, dejará seguramente secuelas a nivel intelectual.13
Continuaremos con un control estricto sobre todo a nivel cognitivo, los resultados de la primera evaluación neuropsicológica no mostraron datos de regresión del neurodesarrollo en sus diversas esferas (lenguaje, motrices, ejecutivas, memoria), ni alteraciones a nivel del lenguaje, solo concomitantemente un TDAH tipo combinado (hiperactivo-inatento), es importante dado el contexto del caso que su medición basal se encuentre dentro de parámetros normales para la edad, por lo que repetirlo cada seis meses o de manera anual, por el momento no logramos completar su diagnóstico definitivo.
Se indicará iniciar además del tratamiento anticomicial, el manejo conjunto con rehabilitación del lenguaje, si es síndrome de Landau Kleffner o terapia educacional de apoyo en el caso del síndrome CSWS, pues sólo un 25% de los pacientes con estas afecciones llegan a ser funcionales para las competencias de la vida adulta.12 La evolución del caso es importante en su seguimiento, pues como ocurre en ciertos padecimientos de la neurología pediátrica sólo el tiempo marcará la pauta de nuestro manejo.
Recibido: Octubre 2011. Aceptado: Febrero 2012
Correspondencia:
Dr. Humberto Iván Ortega García,
Departamento de Neurología Pediátrica, Facultad de Medicina y Hospital Universitario Dr. José Eleuterio González de la UANL.
Av. Francisco I. Madero y Gonzalitos s/n,
Colonia Mitras Centro,
C.P.64460. Monterrey, N. L., México.
Correo electrónico:dr_humberto_ortega@yahoo.com.mx.