



Systemic Lupus Erythematosus (SLE) is a multisystemic autoimmune disease of unknown origin with a waxing and waning course and a significant morbi-mortality. The objective of this paper is to provide an SLE overview, as well as recommendations regarding diagnosis and therapeutic concepts. In the first stage of the disease, the combination of genetic, gender and environmental factors culminate in the formation of autoantibodies years before the onset of symptoms is observed. In the second phase, there are clinical manifestations and associations with comorbidities. Management of patients with SLE should be predictive, preventive, personalized, and participatory in order to achieve remission and prevent relapses. We can divide SLE into three categories according to the severity of the disease: mild, moderate, and severe. Corticosteroids are the mainstay of therapy, but the use of another agent is mandatory in order to reduce side effects. Some of the biological agents used in immunosuppressive therapy in SLE treatment include methotrexate, antimalarials, azathioprine, mycophenolate mofetil, cyclophosphamide, belimumab and rituximab.
BackgroundDiagnosing Systemic Lupus Erythematosus (SLE) has been a challenge over the years. The first reports of the disease only considered skin manifestations. Later, William Osler recognized the systemic involvement of the disease.1 SLE is a multisystemic autoimmune disease of unknown origin.2 SLE has an incidence of 1–10 per 100,000 person-years and a prevalence of 20–70 per 100,000 inhabitants.3 SLE prevalence in Hispanics is 138.7–244.5 per 100,000 people.4 For every 9–10 women with SLE, 1 male will be affected.2 SLE has a waxing and waning course with significant morbidity that can be fatal – if not treated early – in some patients. A diagnosis of SLE should be considered when a patient has characteristic features of SLE associated with autoantibody formation5; thus, the presence of anti-nuclear antibodies (ANA) is considered necessary for an SLE diagnosis. Patients without ANA will have less than a 3% probability of developing the disease.
The objectives of this paper are to provide an overview based on the literature and on the personal experience of 30 years of treating patients with SLE, provide general and specific recommendations regarding the diagnosis of this challenging disease, and share therapeutic concepts that are fundamental for the comprehensive management of the disease.
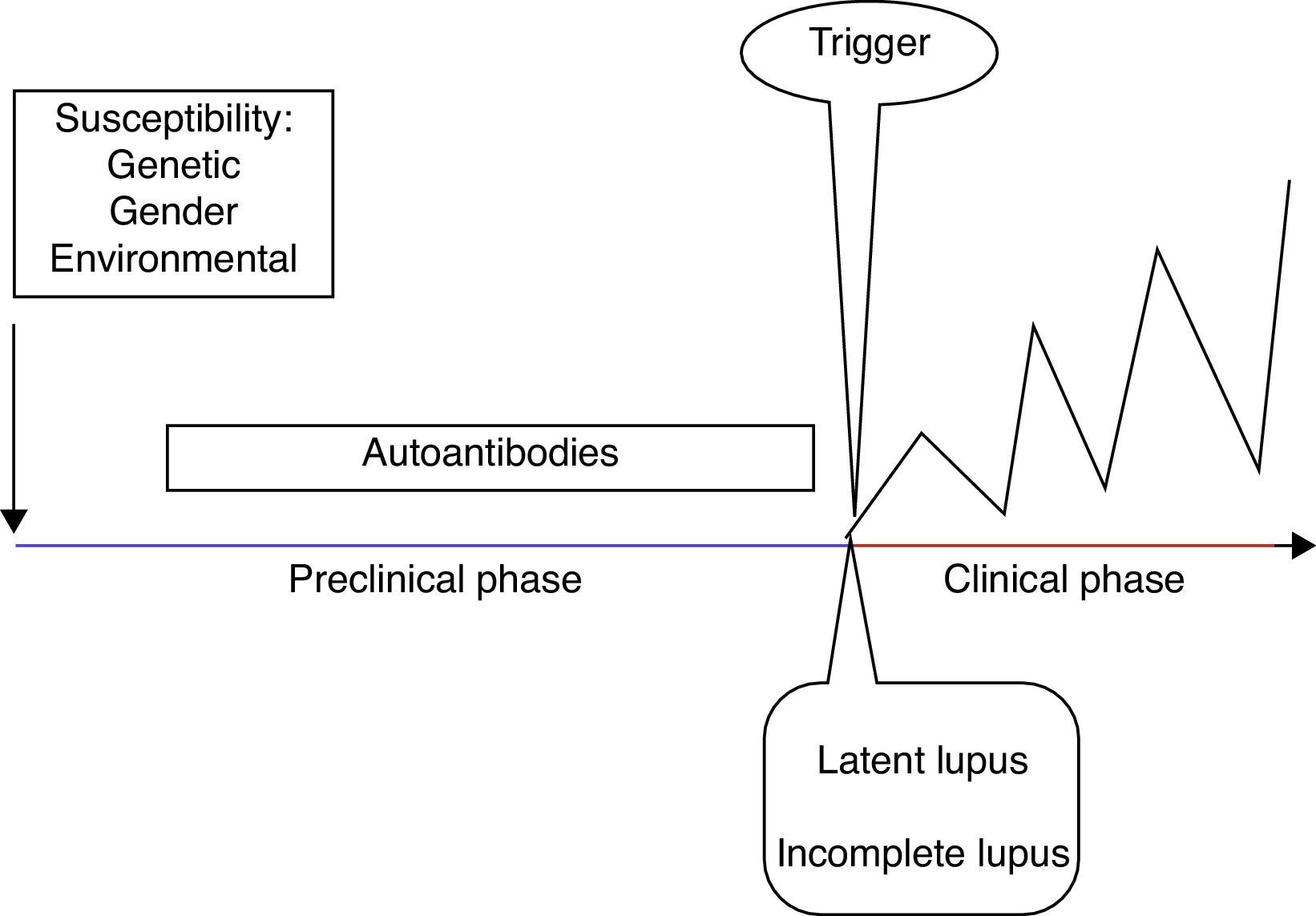
SLE stagesSLE stages include a preclinical and a clinical phase, as well as its related comorbidities.
Clinical manifestations only develop in predisposed individuals and are secondary to a loss of tolerance with a subsequent immune dysregulation6 (Fig. 1). The development of autoimmunity is determined by genetic, gender, and environmental factors. Advances in genetic techniques have identified more than 30 genetic associations with SLE including variants of HLA and Fcγ receptor genes, IRF5, STAT4, PTPN22, TNFAIP3, BLK, BANK1, TNFSF4 and ITGAM.7 Moreover, the genetic contribution to the development of SLE has been observed in twins, with a concordance between monozygotic twins of 24–56% vs 2–5% in dizygotic twins.8 Female preponderance in the pathogenesis of SLE has been demonstrated in transgenic mice. Smith-Bouvier et al. observed that mice with the XX chromosome were more susceptible to developing lupus when compared to XY mice.9 Environmental factors can contribute to the development of SLE by the inhibition of DNA methylation.10 These factors include drugs (e.g. procainamide), diet, smoking, UV light exposure and infections (Epstein–Barr virus).11 Finally, there is a pathogenic autoantibody production in SLE patients, reflecting loss of tolerance.6

Different authors have described the development of autoantibodies before the clinical onset of the disease in the past. Arbuckle et al. described the presence of at least one SLE autoantibody before the diagnosis (up to 9.4 years earlier; mean, 3.3 years) in asymptomatic patients. Antinuclear, antiphospholipid, anti-Ro and anti-La antibodies preceded the other autoantibodies in this cohort of patients.12 Subsequently, McClain et al. described the clinical significance of the presence of antiphospholipid antibodies prior to an SLE diagnosis, as well as the presence of these autoantibodies in patients with a more severe clinical outcome.13
In order to classify patients in the early stages of the disease, different authors have proposed definitions according to the symptoms and the presence of classification criteria. First, the term undifferentiated connective tissue disease (UCTD) is used in individuals with a disease manifestation suggestive but not diagnostic of a specific connective tissue disease. UCTD accounts for 10–20% of referred patients, 10–15% will fulfill the classification criteria for SLE 5 years later.14 Factors that predict evolution to SLE are young age, alopecia, serositis, discoid lupus, a positive anti-human globulin (Coombs) test and anti-Sm or anti-DNA antibodies.15
Ganczarczyk et al. described the term “latent lupus” to define patients with features consistent with SLE which may or may not be a part of the American College of Rheumatology (ACR) classification criteria, but still are ≤4.16
Incomplete lupus refers to patients with less than four ACR classification criteria for SLE. Swaak et al. in a multicentric study, observed that only three of 122 incomplete lupus patients developed SLE during 3 years of follow-up, and suggested that incomplete SLE forms a subgroup with a good prognosis.17 Later, Greer et al. confirmed this observation. They followed 38 incomplete lupus patients over 19 months and only two developed SLE.18 An additional term is preclinical lupus, which defines individuals with increased genetic risk for the development of SLE but no clinical symptoms.19
After the preclinical stage, the clinical stage occurs with the onset of symptoms. The GLADEL (Grupo Latinoamericano de Estudio de Lupus) cohort, a multinational inception prospective cohort in Latin American centers, described the symptoms in 1214 patients with SLE. They found that arthralgia and/or arthritis, fever, photosensitivity, alopecia and malar rash were the most common symptoms at onset.20
SLE treatmentSLE management represents the “P4”, a new paradigm of modern medicine. P4 Medicine stands for Predictive, Preventive, Personalized and Participatory Medicine.
SLE is a syndrome with high variability in the disease course as well as in the severity of the manifestations; therefore each SLE patient should be treated on an individualized basis in order to implement a proper treatment.21 The goal of the treatment is to achieve remission, prevent flares and use of drugs with the minimum dose required to prevent long-term side effects. The treatment includes lifestyle modification, patient education, physical activity and medical or (in some cases) surgical intervention.
There are general recommendations that are given to SLE patients (Table 1). All patients should have a balanced diet and exercise regularly. Patients are advised to avoid Echinacea, melatonin, garlic, and alfalfa sprouts, which have been described to precipitate their condition.22 It is also important to inform patients to avoid disease reactivation drugs such as procainamide, hydralazine, sulfonamides, anti-TNFa, ibuprofen or estrogen.23,24 Smoking also appears to influence the onset and course of the disease among patients with SLE.25,26 The effect of drugs like methotrexate (MTX) and hydroxychloroquine (HCQ) may diminish with smoking.
General recommendations for SLE* patients.
| Balanced diet and exercise |
| Avoid substances and drugs that might induce lupus |
| No smoking |
| Vaccination schedule |
| Assessment of cardiovascular risk factors |
| Screening of cancer |
| Evaluation of reproductive health |
| Assessment of cognitive function |
The vaccination schedule in SLE patients includes a yearly influenza vaccine and a pneumococcal vaccine every 5 years. Hepatitis B and Tetanus toxoid vaccinations also seem to be safe, and not associated with flares.27 The quadrivalent human papillomavirus vaccine is also safe and not associated with increased lupus activity.28 It is important to consider that inactivated live vaccines are contraindicated in patients taking immunosuppressive drugs and/or glucocorticoids at a dose >20mg/day.27
Most SLE patients are diagnosed in the reproductive years, thus reproductive health is an important issue. It is recommended for SLE patients to have an inactive disease for a six month period prior to conception. There are three main types of contraceptives: barrier methods, intrauterine devices and the hormonal method. Hormonal methods include combined or progestin-only. The use of combined methods is associated with an increased risk of SLE,29 however, progesterone methods have proven to be safe for SLE patients.30
In addition to the control of the disease, SLE patients should have a systematic assessment of comorbidities. SLE patients develop premature atherosclerosis and their risk of heart attack and stroke is 10 times higher than that of age-matched controls.31 Atherosclerosis is the result of the complex interplay between dysfunctional immune regulation, inflammation, traditional risk factors, aberrant endothelial cell function and repair, and the therapeutics for treating the underlying autoimmune disease.32 SLE patients also have an increased risk of different types of cancer such as non-Hodgkin lymphoma, lung cancer, and cervical dysplasia. Lupus disease activity, smoking and immunosuppressive drug exposure are some of the causes of the increase in cancer risk.33 Therefore, SLE patients should have colonoscopies, Pap smear, and mammogram schedules.
Cognitive dysfunction prevalence in SLE ranges from 12% to 87%. Petri et al. compared cognitive functioning in recently diagnosed SLE patients versus normal controls. Using Automated Neuropsychological Assessment Metrics (ANAM), SLE patients performed significantly worse than normal controls. Therefore, a cognitive assessment is necessary in all SLE patients from the onset of the disease.
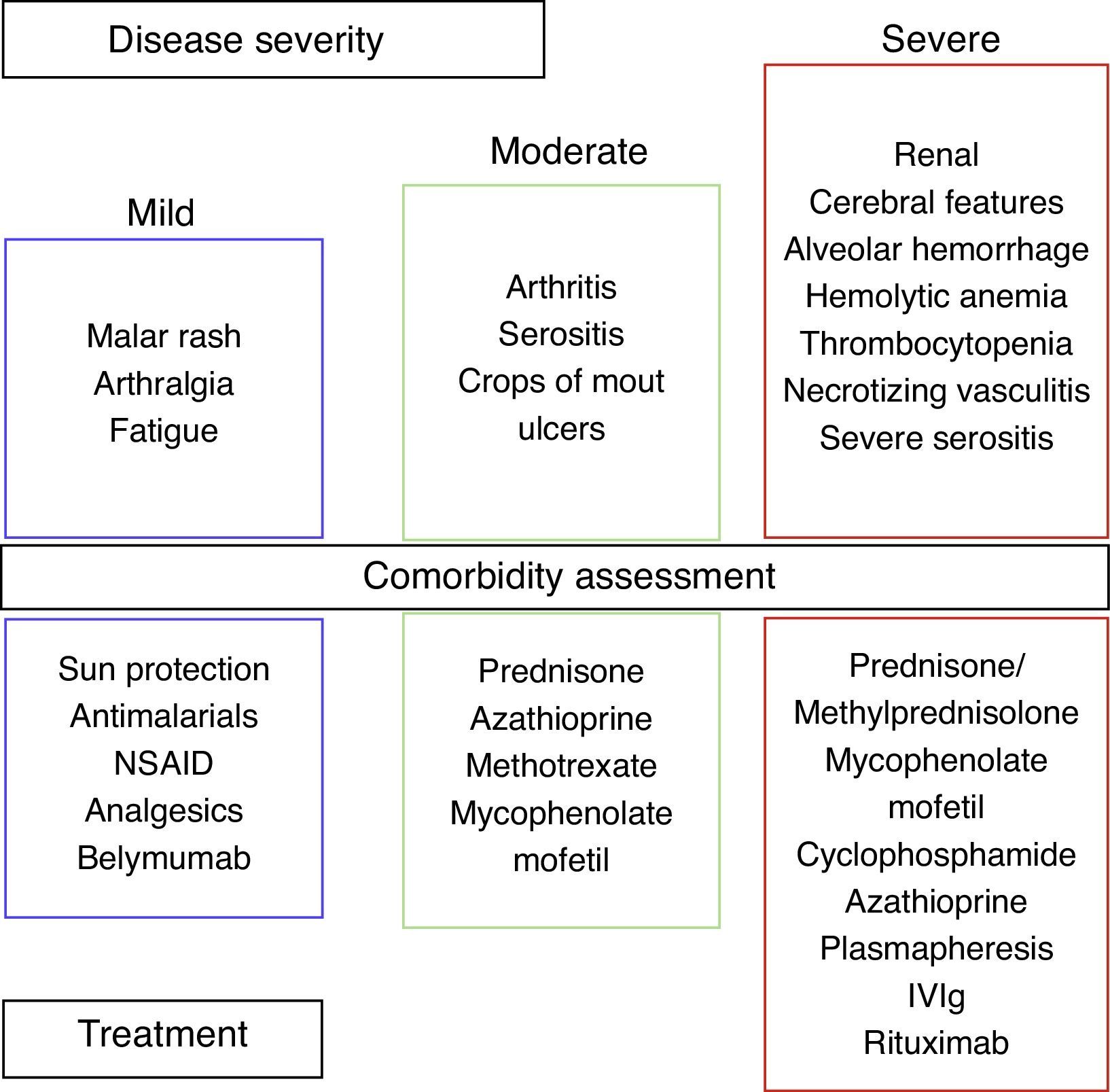
We can divide SLE into three categories based on disease severity: mild, moderate, and severe (Fig. 2).

Corticosteroids (CS) are the mainstay of treatment for SLE in any category, with proven efficacy.34 The dose varies according to the severity of symptoms. A low dose is 0.1–0.2mg/kg/day, an intermediate dose is 0.3–0.5mg/kg/day, and a high dose is 0.6–2mg/kg/day. The use of this drug is associated with an increase in serum lipids, blood pressure, weight and glucose, in addition to cataracts and osteoporotic fractures. The adverse side effects of CS depend on both the current and the cumulative dosage. Thamer et al. demonstrated the hazard ratio for accrued organ damage to be 1.5, 1.64 and 2.51 with prednisone doses of 6mg/day, 12mg/day and >18mg/day, respectively.35 It is important to note that SLE diagnosis is not equivalent to the use of methylprednisolone, and that in many cases the deleterious effects of CS may outweigh the benefits. Therefore, the goal is the use of CS according to the clinical manifestations and slow tapering to 1–2mg/day. In order to reduce CS doses and side effects, the use of another agent is mandatory.34
Mild SLE includes mucocutaneous lesions, arthralgias and fatigue. Sun protection consists of avoiding when the sun is at its highest (10am to 4pm) and patients should use agents with a sun protection factor of at least 50, applied 20–30min prior to exposure, and reapplied every 4h. Topical therapies depend on whether it is a localized or widespread skin disease. Therapies include steroids and/or calcineurin inhibitors.36 Systemic therapies include antimalarial agents, MTX, azathioprine, mycophenolate mofetil (MMF), dapsone, and cyclophosphamide (CYC), and are used in refractory diseases or in poor responses to treatment.34,36 For cutaneovascular manifestations (Raynaud syndrome, livedo reticularis, etc.) the use of cold-preventive measures and calcium channel blockers can be beneficial. Nonsteroidal antiinflammatory drugs (NSAID) can be used in headaches, myalgias, arthralgias, and serositis. NSAID use must be monitored; side effects could be renal, gastrointestinal or cardiovascular. In my experience, I have seen severe secondary side effects such as aseptic meningitis. Ibuprofen is the drug most frequently involved in aseptic meningitis, but sulindac and naproxen have also been described.37
Moderate SLE includes arthritis, serositis, and crops of mouth ulcers. Arthritis can improve with NSAID, moderate doses of prednisone, or antimalarial drugs. When the response is poor, MTX, leflunomide, azathioprine and TNF-a agents can be used.38 Serositis (pleurisy, pericarditis) responds to NSAID and CS. Belimumab is a fully humanized IgG1 mAb that binds to soluble BLyS (B lymphocyte stimulator), inhibiting its activity.21 BLISS-52 and BLISS 76 demonstrated significant clinical responses with Belimumab compared to placebos in patients with mild and moderate disease activity (without nephritis/CNS).39,40
The severe SLE stage includes hemolytic anemia, thrombocytopenia, diffuse alveolar hemorrhage, necrotizing vasculitis, neuropsychiatric lupus and renal involvement. In this stage, CS is used in high doses and intravenous methylprednisolone pulses for severe cases. In hemolytic anemia and thrombocytopenia the treatment includes CS and danazol, Rituximab, intravenous immunoglobulin (IVIg), MMF, CYC, plasmapheresis and/or splenectomy for refractory cases.41 The use of CYC and high doses of CS are also employed for diffuse alveolar hemorrhage, and plasmapheresis for refractory cases. We made an observational, retrospective study that included twelve SLE patients with alveolar hemorrhage. We found that simultaneous treatment with CS, CYC, plasmapheresis and IVIg was associated with a mortality of 17%, contrary to the rate previously described of up to 70–90%.42
According to the recommendations, glucocorticoid and immunosuppressive therapy is indicated for severe neuropsychiatric manifestations (myelopathy, optic neuritis, etc.). Anticoagulation therapy is indicated for the SNC manifestations of antiphospholipid syndrome.43 In our practice, we have also observed that the combination of methylprednisolone, CYC, IVIg and Rituximab was effective for psychosis refractory to conventional treatment.
Renal involvement is considered the most important prognostic factor. The Task Force Panel for screening, treatment, and management of Lupus Nephritis (LN) recommended the treatment to be based on the type of LN according to the ISN/RPS criteria.44 The treatment consists of the use of corticosteroids either solely or in combination with immunosuppressive agents. The recommendations for LN treatment include an induction and a maintaining therapy. There are 2 regimens for Class III/IV LN, low-dose “Euro-Lupus” CYC and high-dose CYC followed by maintenance treatment with MMF or azathioprine.45 In our practice, we believe that a low-dose CYC is more beneficial to patients decreasing adverse effects, such as infections, gonadal toxicity and increased risk of cancer. We do not share the idea that methylprednisolone pulses will provide a greater benefit than prednisone. And finally, LN response should be evaluated 3–6 months after initiating treatment.
In conclusion, SLE is a challenging disease to diagnose and treat. Advances in research have allowed us to know which individuals are at risk of developing the disease. Each patient should be treated on an individualized basis according to their clinical manifestations in order to provide proper treatment.
FundingNo financial support was provided.
Conflict of interestThe authors have no conflicts of interest to declare.