


La amiloidosis sistémica es una enfermedad ocasionada por el depósito de una proteína fibrilar en diversos órganos.
ObjetivoDocumentar el papel de la biopsia renal en la determinación del tipo de amiloidosis en un cuadro con presentación atípica y sospecha de amiloidosis secundaria a psoriasis.
MétodoMasculino de 64 años, con antecedente de enfermedad renal crónica de etología no afiliada en 2 hermanos. Tiene antecedentes personales de lesiones dérmicas escamosas eritematosas de más de 10 años sin diagnóstico. Presenta edema en extremidades inferiores y proteinuria de rango subnefrótico.
ResultadosSe realiza biopsia renal, reportándose amiloidosis renal.
ConclusionesLa biopsia renal es una herramienta fundamental para patologías renales, tanto para el diagnóstico como para el pronóstico del paciente.
Systemic amyloidosis is a disease caused by the deposition of a fibrillar protein in various organs.
ObjectiveTo document the role of renal biopsy in determining the type of amyloidosis in a clinical picture with atypical presentation and suspicion of amyloidosis secondary to psoriasis.
MethodA 64-year-old male with a history of chronic renal disease of unknown origin in two siblings. She has a personal history of undiagnosed erythematous squamous skin lesions of more than 10 years onset. She had swelling in the lower limbs, with urine protein in the sub-nephrotic range proteinuria.
ResultsA renal biopsy was performed, which was reported as renal amyloidosis.
ConclusionsRenal biopsy is a fundamental tool for investigating renal disorders both for diagnosis and for patient prognosis.
La amiloidosis sistémica es una enfermedad ocasionada por el depósito de una proteína fibrilar en diversos órganos. Se diferencian entre sí por el tipo de proteína precursora de la sustancia amiloide. Cabe señalar que la inmunohistoquímica es una técnica que permite clasificar el tipo de amiloide y se realiza rutinariamente. Una amiloidosis primaria con demostración de rojo Congo positivo y ninguna evidencia de proliferación clonal de las células plasmáticas debe obligar a descartar otras formas de amiloidosis, en particular la amiloidosis familiar, y más si tiene antecedentes familiares de enfermedad renal de etiología desconocida. El diagnóstico diferencial es complicado, pero es relevante para el pronóstico y el tratamiento.
ObjetivoDocumentar el papel de la biopsia renal (BR) en la determinación del tipo de amiloidosis en un cuadro con presentación atípica y sospecha de amiloidosis secundaria a psoriasis.
MétodoMasculino de 64 años, con antecedentes de enfermedad renal crónica de etología no filiada en 2 hermanos de la sexta década de la vida. Cuenta con antecedente personal de lesiones dérmicas eritematosas descamativas de más de 10 años de evolución, sin diagnóstico establecido. Presenta STDA secundario a úlcera duodenal, con hospitalización por 2 semanas; curso con lesión renal aguda con creatinina sérica al egreso de 1,6mg/dl. Dos meses después, con edema de miembros pélvicos e hiperbromuria, por lo que se refiere a nuestro centro médico para realización de BR.
En cuanto a laboratorios de extensión, se reportó lo siguiente: hemoglobina 13,4 g/dl, leucocitos 5,5 millones/mm3, creatinina sérica 1,50mg/dl, nitrógeno ureico en la sangre 27mg/dl, sodio 140 mEq/l, potasio 5,6 mEq/l, calcio 8,7mg/dl, fosfatasa alcalina 155 U/l, bilirrubina total 0,80mg/dl, colesterol total 184mg/dl, albúmina 3,2 g/dl, proteinuria en 24h de 1,2 g en 24h y albuminuria en 24h 489mg. El examen general de orina reporta densidad de 1.015, pH 5, color amarillo, leucocitos negativos, nitritos negativos, proteínas ++, glucosa negativo, eritrocitos+.
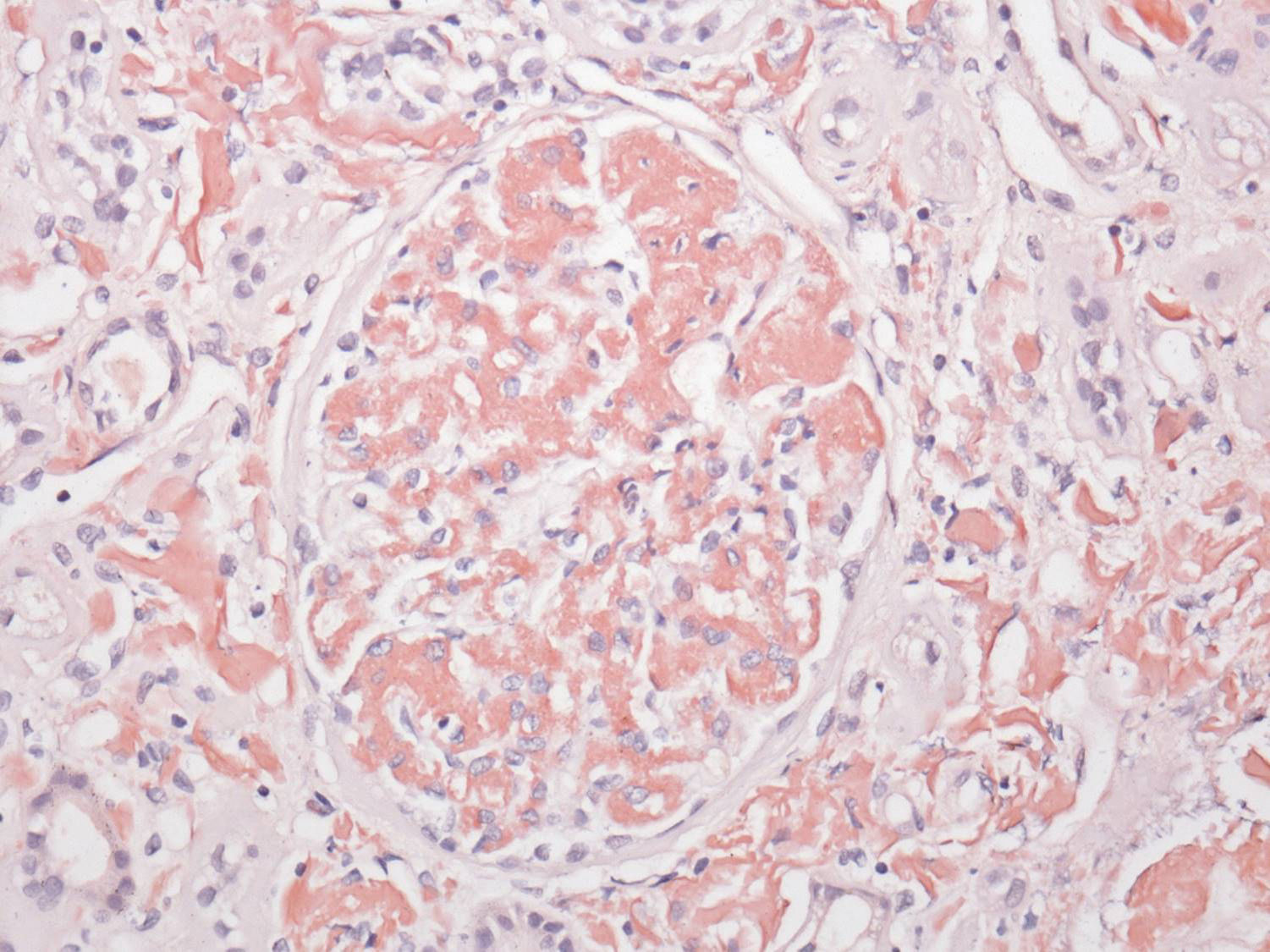
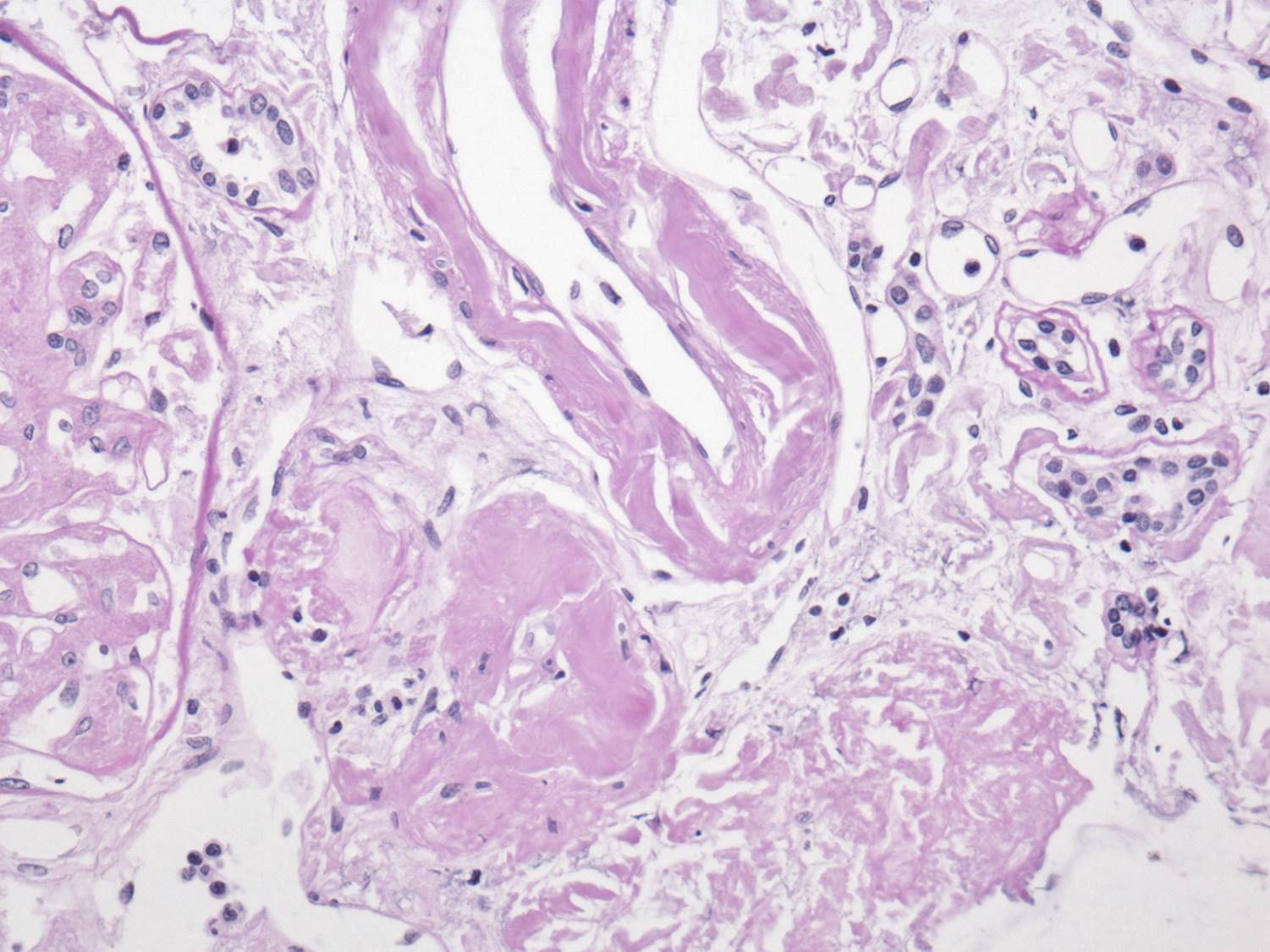
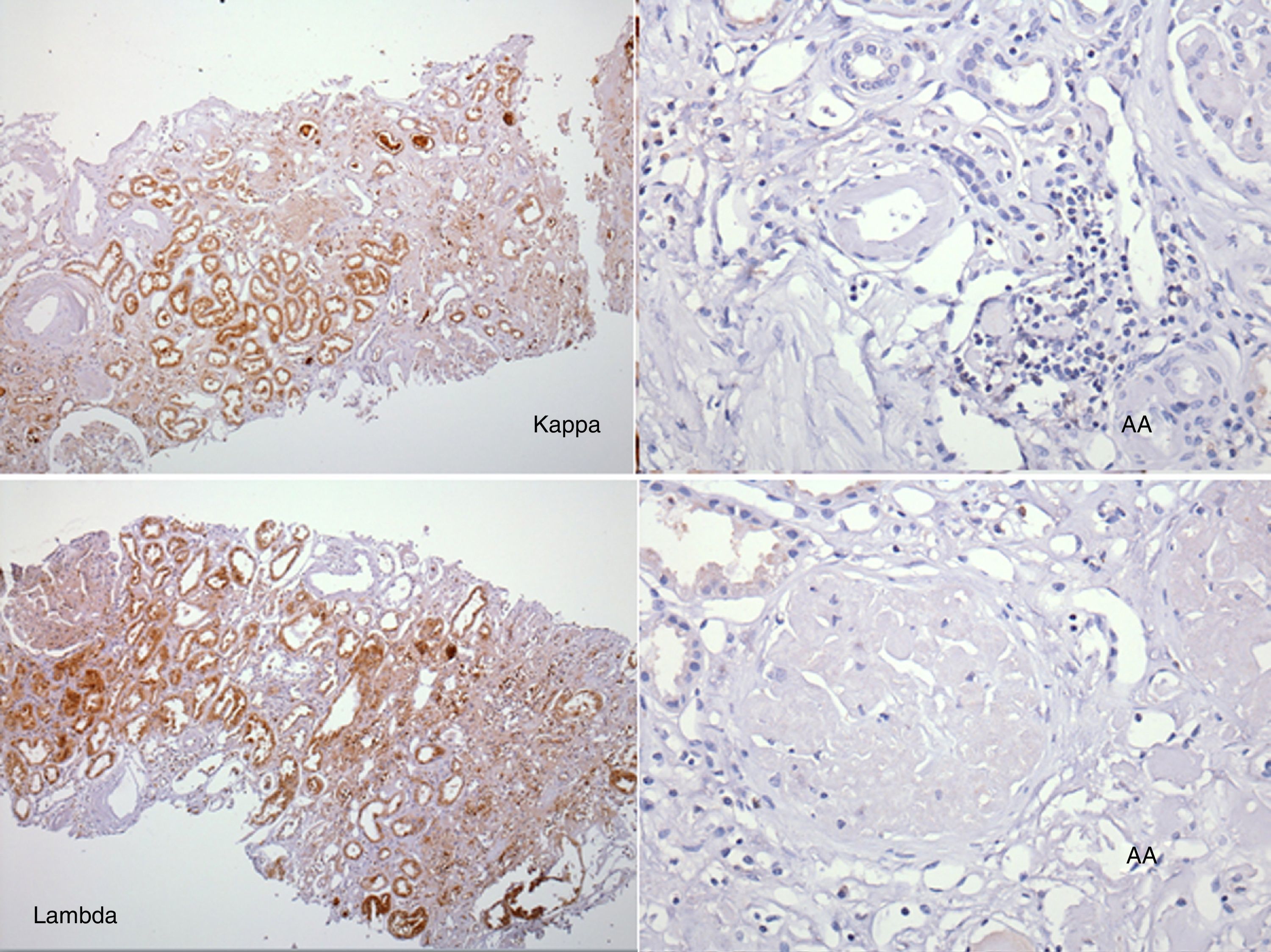
El perfil viral, sin alteraciones. La determinación de inmunoglobulinas con IgG 1.900mg/dl; el resto, normales. Cadenas ligeras séricas sin picos monoclonales y cadenas ligeras en orina normales. Biopsia de hueso sin patología aparente. El reporte del USG renal con riñón derecho 12×7×6,7cm y riñón izquierdo de 11×6,5×5,3cm. Tomografía computarizada abdominal sin evidencia de megalias ni lesiones infiltrativas, así como panendoscopia con varices esofágicas, gastropatía hipertensiva y duodenitis erosiva. La biopsia de piel diagnosticó psoriasis. La BR con reporte histopatológico de amiloidosis glomerular (fig. 1: la tinción de rojo Congo [para amiloide] resultó positiva), intersticial y vascular con cambios regenerativos focales de epitelio tubular (fig. 2: el material amiloide también se encuentra depositado en el intersticio, en las paredes tubulares y arteriolares). Se efectúa marcación para amiloide AA, reportándose negativa (fig. 3: se efectuó marcación para amiloide AA, kappa y lambda a través de la técnica de inmunoperoxidasa indirecta en el tejido del bloque de parafina, reportándose negativa).



Amiloidosis es un término genérico para un grupo de enfermedades que son causadas por el plegamiento erróneo y la acumulación extracelular de varias proteínas. Estas proteínas plegadas erróneamente forman depósitos fibrilares que producen birrefringencia verde manzana patognomónica cuando se tiñen con colorante rojo Congo y se ven bajo luz polarizada usando microscopia [1].
Se estima que la incidencia ajustada por edad de la amiloidosis AL en Estados Unidos y en el Reino Unido es de 5,1-12,8 casos por millón de personas por año. La enfermedad se desarrolla en aproximadamente el 2% de los individuos con discrasias monoclonales de células B [2].
Se han descrito otras formas de amiloidosis familiar y se han identificado sus proteínas anómalas precursoras junto con sus mutaciones asociadas tales como amiloidosis renal, hepática y polineuropática en los EE. UU. y Reino Unido, debido a una variante anómala de la apoproteína AI [3].
La amiloidosis secundaria es comúnmente asociada con una variedad de trastornos inflamatorios crónicos, tales como artritis reumatoide, espondilitis anquilosante y osteomielitis. En este caso clínico, se tiene el diagnostico de psoriasis como factor de riesgo para una forma secundaria de amiloidosis, sin embargo, rara vez se ha informado estar asociado con la psoriasis. En un estudio de la Clínica Mayo se reportaron 28 casos de enfermedad coexistente con amiloidosis en un periodo de 42 años, y en 5 de estos casos la psoriasis fue la única causa que precede al desarrollo de la amiloidosis secundaria [4].
La amiloidosis familiar se hereda principalmente como trastorno autosómico dominante. El tipo más común de amiloidosis familiar es amiloidosis transtiretina, pero podemos reconocer otros tipos con proteínas que incluyen la cadena de A de fibrinógeno (AFib), lisozima (ALys), apolipoproteínas AI (AApo AI) y AII (AApo AII) y gelsolina (AGel) [5].
Recientemente, se ha descrito un nuevo tipo de amiloidosis altamente prevalente en la población hispana derivado del factor quimiotáctico de los leucocitos 2 (LECT2) [6]. La mayoría de los casos notificados se han descrito en pacientes de ascendencia mexicana, por lo cual se puede argumentar un papel genético [7].
Las manifestaciones clínicas relacionadas con ALECT2 se han limitado en gran medida al riñón; sin embargo, pudiera haber depósitos en tejido hepático, esplénico, colónico y adrenal [8].
Este tipo de los pacientes presentan diferentes grados de deterioro de la función renal. La proteinuria es sorprendentemente baja en comparación con otras formas de amiloidosis [9].
En nuestro caso clínico, no se debe descartar una forma hereditaria o por LECT2, ya que presenta antecedentes familiares de 2 hermanos con enfermedad renal crónica de etiología no filiada iniciada en sexta década de la vida, así como una presentación clínica atípica con una proteinuria subnefrótica con una tinción en BR para AA negativa.
La importancia de un correcto diagnóstico diferencial en la amiloidosis sistémica radica en que el manejo y el pronóstico de la enfermedad pueden ser completamente diferentes según sea el origen de la misma, para lo cual tiene especial relevancia determinar la proteína que origina el depósito [10].
Entre los métodos disponibles para determinar la composición proteica de las fibrillas se encuentran la inmunohistoquímica y la secuenciación directa de la proteína. Para realizar esta última técnica se requiere una cantidad considerable de tejido, algo que no siempre es posible obtener [11].
La microdisección láser y el análisis proteómico basado en espectrometría de masas (LMD/MS) han demostrado ser una herramienta precisa y útil en la tipificación de amiloide [12].
Se recomienda la identificación de amiloide basada en espectrometría de masas y microdisección con láser en los casos en que la inmunofluorescencia de rutina y/o las pruebas inmunohistoquímicas no pueden tipificar definitivamente los depósitos amiloides [12,13].
ConclusiónEn este caso, además de orientar al diagnóstico, la BR nos ayudó a clasificarla como una potencial tipo familiar al documentarse tinción AA negativa, junto a la ausencia de cadenas libres y descartar la asociación de la psoriasis como forma secundaria.
Responsabilidades éticasProtección de personas y animalesLos autores declaran que para esta investigación no se han realizado experimentos en seres humanos ni en animales.
Confidencialidad de los datosLos autores declaran que han seguido los protocolos de su centro de trabajo sobre la publicación de datos de pacientes.
Derecho a la privacidad y consentimiento informadoLos autores han obtenido el consentimiento informado de los pacientes y/o sujetos referidos en el artículo. Este documento obra en poder del autor de correspondencia.
Conflicto de interesesEn este artículo no existe conflicto de intereses.