



La deficiencia cerebral de folato (DCF) es un síndrome neurológico asociado con una baja concentración de 5-metiltetrahidrofolato (5MTHF) en el líquido cefalorraquídeo (LCR) en presencia de folato periférico normal1. El síndrome clínico se puede encontrar desde el período prenatal hasta la edad adulta, con una amplia gama de fenotipos2. La generación de autoanticuerpos contra el receptor de folato3, las variantes patógenas en el gen receptor de folato 1 (FOLR1) y la disfunción mitocondrial han sido reportados como causas de DCF2,4. Sin embargo, es probable que otras causas estén involucradas, ya que la DCF fue reportada en una variedad de desórdenes neurológicos y psiquiátricos5,6.
Presentamos el caso de un paciente con DCF que luego fue diagnosticado de paraplejía espástica hereditaria.
Un hombre de 26 años desarrolló, a la edad de 10 años, un cuadro clínico progresivo de trastorno cognitivo-conductual, dificultades para caminar y un temblor generalizado con inicio en el miembro superior izquierdo.
Cuando se le observó por primera vez, a la edad de 16 años, presentó una implicación neurológica multifocal con regresión cognitiva, movimientos oculares intrusivos, un trastorno del movimiento (distonía multifocal, temblor generalizado y bradiquinesia), un síndrome piramidal y ataxia de la marcha con pie cavo.
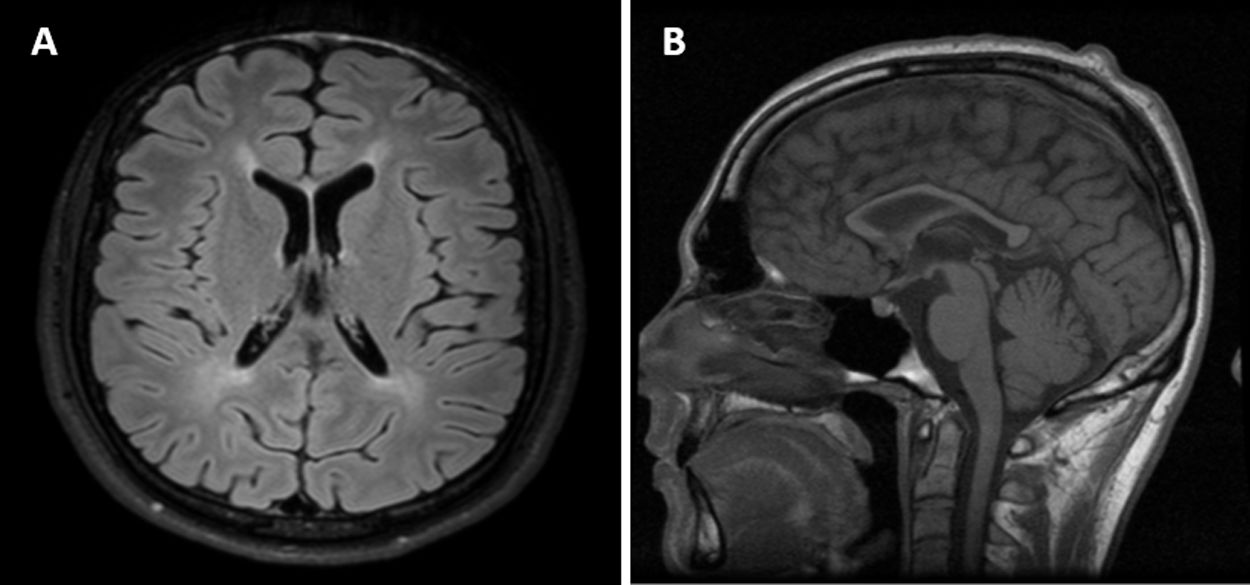
La evaluación neuropsicológica reveló una disfunción frontoestriatal y el electroencefalograma tenía una actividad basal lenta (6Hz). La resonancia magnética (RM) del cerebro mostró una hiperintensidad de la materia blanca periventricular y una atrofia global con una atrofia desproporcionada del cuerpo calloso (fig. 1).
 y la atrofia global con atrofia desproporcionada del cuerpo calloso en sagital T1 (B).")
El paciente fue tratado con levodopa, con una notable respuesta del síndrome extrapiramidal.
Posteriormente, se identificó una DCF (folato del LCR < 20 nmol/l; rango normal 50-78 nmol/l), con niveles normales de neurotransmisores, pterinas y aminoácidos. El folato sérico estaba normal. Se excluyeron las variantes en los genes FOLR1 y POLG, y en el ADN mitocondrial. El análisis de los anticuerpos contra el receptor de folato en suero mostró un título aumentado de los anticuerpos bloqueadores (4,77pmol folato bloqueado/ml de suero).
La suplementación con ácido folínico (30mg/día) se inició, con la normalización de los niveles en el LCR y la mejora significativa de la disfunción cognitiva, las anomalías extrapiramidales, oculomotoras y electroencefalográficas, sin mejorar los signos piramidales.
A los 18 años, era evidente una paraparesia espástica. Hoy en día, no puede estar de pie o caminar sin ayuda. Otros signos neurológicos, como el parkinsonismo y la disfunción cognitiva, a pesar de la reducción progresiva de la levodopa y de los agentes anticolinérgicos, prácticamente desaparecieron. La RM de cerebro y de la médula espinal excluyó nuevas lesiones estructurales y el 5MTHF en el LCR se mantuvo normal. El paciente también realizó 3estudios electrofisiológicos, a los 19 y 24 años, que excluyeron polineuropatía o miopatía.
Recientemente, la secuenciación del exoma clínico identificó 2variantes en el gen ZFYVE26, c.1675T>C (p.(Cys559Arg)) y c.3394C>T (p.(Gln1132*)), y una variante en el gen BSCL2, c.1220C>T (p.(Pro407Leu)). La variante c.3394C>T (p.(Gln1132*)) en el gen ZFYVE26 nunca había sido reportada y resulta en una proteína truncada, siendo clasificada como patógena. La otra variante también es nueva, pero de importancia clínica desconocida, como es el caso de la variante identificada en el gen BSCL2. El estudio de los padres asintomáticos mostró que las 2variantes de ZFYVE26 están en alelos diferentes; la variante del gen BSCL2 también se encontró en el padre sano del paciente. Después de este hallazgo, el paciente siguió recibiendo suplementos de ácido folínico.
Se especula que los diferentes fenotipos de la DCF están determinados por la edad en la que la transferencia de folato del sistema nervioso central se ve afectada.2. La distonía localizada que luego se generaliza, con bradiquinesia y síndrome piramidal, se ha descrito en la DCF que se presenta desde la adolescencia hasta la edad adulta2. Otras características incluyen disfunción cognitiva y ataxia de la marcha2. Esta descripción parece ser consistente con el cuadro clínico de nuestro paciente.
La detección de títulos aumentados de anticuerpos antifolato refuerza la asociación entre la DCF y su trastorno. Cabe señalar que se ha detectado la presencia de anticuerpos bloqueadores en varias afecciones, como el autismo, la esquizofrenia, el síndrome de Rett y la enfermedad de Alpers6,7.
La progresión del síndrome piramidal justificó la solicitud de un exoma clínico que permitió la identificación de variantes en el ZFYVE26 y BSCL2, asociadas con la paraplejía espástica autosómica recesiva tipo 15 (SPG15) y la paraplejía espástica autosómica dominante tipo 17 (SPG17), respectivamente. El fenotipo clínico de este paciente evoca características de la SPG15: deterioro cognitivo y manifestaciones conductuales, pies cavos, ataxia, hiperintensidades de materia blanca cerebral en la RM, cuerpo calloso delgado y edad de inicio entre 5 y 19 años8. Aunque la variante BSCL2 estaba presente en el padre asintomático del paciente y este gen se asocia con variabilidad fenotípica y penetrancia incompleta9, el cuadro clínico no está muy en consonancia con la SPG17 y la variante tiene un significado incierto.
Al igual que otras enfermedades, la DCF podría estar asociada con la SPG15 y es probablemente responsable de parte de la condición de este paciente, considerando la respuesta positiva a la suplementación de ácido folínico, especialmente en las características extrapiramidales. Hasta donde sabemos, esta asociación no ha sido descrita hasta ahora, a nivel clínico o molecular. Nos preguntamos si algunos rasgos vinculados a la SPG15 en informes anteriores, a saber, el parkinsonismo10, podrían estar conectados con la DCF. Este caso nos lleva a considerar una posible conexión entre estas 2entidades y la necesidad de ponderar la investigación de la DCF en este tipo de enfermedades.
FinanciaciónLos autores declaran no haber recibido financiación para la realización de este trabajo.
Agradecemos al paciente y su familia.
Agradecemos también la colaboración de Laura Vilarinho y Vasco Sá Pinto en la investigación del paciente.

