





Duchenne muscular dystrophy (DMD) is the most common myopathy in children, with a worldwide prevalence of approximately 0.5 cases per 10000 male births. It is characterised by a progressive muscular weakness manifesting in early childhood, with the subsequent appearance of musculoskeletal, respiratory, and cardiac complications, causing disability, dependence, and premature death. Currently, DMD is mainly managed with multidisciplinary symptomatic treatment, with favourable results in terms of the progression of the disease. It is therefore crucial to establish clear, up-to-date guidelines enabling early detection, appropriate treatment, and monitoring of possible complications.
DevelopmentWe performed a literature search of the main biomedical databases for articles published in the last 10years in order to obtain an overview of the issues addressed by current guidelines and to identify relevant issues for which no consensus has yet been established. The degree of evidence and level of recommendation of the information obtained were classified and ordered according to the criteria of the American Academy of Neurology.
ConclusionsDMD management should be multidisciplinary and adapted to the patient's profile and the stage of clinical progression. In addition to corticotherapy, treatment targeting gastrointestinal, respiratory, cardiac, and orthopaedic problems, as well as physiotherapy, should be provided with a view to improving patients’ quality of life. Genetic studies play a key role in the management of the disease, both in detecting cases and potential carriers and in characterising the mutation involved and developing new therapies.
La distrofia muscular de Duchenne (DMD) es la miopatía más frecuente en niños, con una prevalencia mundial de aproximadamente 0,5 por cada 10.000 varones. Se caracteriza por una debilidad muscular progresiva al inicio de la infancia con aparición posterior de complicaciones musculoesqueléticas, respiratorias y cardíacas que ocasionan discapacidad, dependencia y muerte prematura. Actualmente su tratamiento se fundamenta en medidas sintomáticas multidisciplinares que han modificado favorablemente el curso de la enfermedad, por lo que resulta crucial establecer unas directrices claras y actualizadas que permitan tanto una detección temprana de la enfermedad como un adecuado tratamiento y seguimiento de sus posibles complicaciones.
DesarrolloCon el fin de obtener una visión general de los aspectos abordados por las guías actuales y detectar aquellos en los que todavía no existe un consenso y su abordaje sea relevante, se realizó una revisión de la literatura en la base de datos biomédicas de los últimos 10años. El grado de evidencia y el nivel de recomendación de la información obtenida se clasificaron y ordenaron de acuerdo con los criterios de la American Academy of Neurology (AAN).
ConclusionesEl abordaje de la DMD debe ser multidisciplinar y ajustado al perfil del paciente y su grado de evolución clínica, comprendiendo, además del tratamiento basado en corticoides, medidas a nivel gastrointestinal, respiratorio, cardiaco, fisioterapéutico y ortopédico dirigidas a mejorar la calidad de vida de los pacientes. Los estudios genéticos desempeñan un papel clave en el manejo de la enfermedad, tanto en la detección de casos y posibles portadoras como en la caracterización de la mutación implicada y el desarrollo de nuevas terapias.
Duchenne muscular dystrophy (DMD) is an X-linked recessive disease affecting one in every 3800-6300 male live births.1 Its worldwide prevalence is approximately 0.5 per 10000 male population,2 which translates into 1000 cases in Spain and 12500 in the European Union, making it the most frequent muscular dystrophy in children. DMD is characterised by progressive muscular weakness with onset in early childhood, later progressing to complications leading to disability, dependence, and premature death.
Most patients are diagnosed between 3 and 5 years of age.3 Muscular weakness progresses until the use of a wheelchair is necessary before adolescence, with respiratory, orthopaedic, and cardiac complications appearing simultaneously or subsequently.4 Neurocognitive dysfunction is also frequent; while this is not progressive,5 it does have an impact on learning capacity and quality of life. In recent years, correct management of complications6,7 and the use of corticosteroids8 have enabled survival to be prolonged until the third or fourth decade of life.9–13
DMD is caused by several mutations of the dystrophin gene (DMD, locus Xp21.2),14 leading to the lack of a subsarcolemmal protein which is essential to the structural stability of the muscle, triggering severe, progressive muscular degeneration.15 There are milder allelic variants, such as Becker muscular dystrophy (BMD), in which dystrophin is only decreased and ability to walk is preserved beyond the age of 16; there are also intermediate variants between DMD and BMD, and further variants affecting only the heart or progressing with no or very few symptoms.
Therapeutic management of DMD is essentially symptomatic, based on a set of protocolised actions, with the aim of improving functionality and quality of life, delaying and treating complications, and improving survival. It is therefore vital to establish clear guidelines to enable both early detection and adequate follow-up of the disease.
ObjectivesAlthough there are clinical consensus guidelines addressing different aspects of DMD management,8,16–18 no updated guidelines including the most recent advances on the disease are available in Spain. The main aim of this document is to assist healthcare professionals in the diagnosis and treatment of the disease and in decision-making regarding all relevant aspects of disease management.
PopulationSince DMD presents an X-linked recessive inheritance pattern, it mainly affects men. However, 10% of female carriers of the mutation show some typical manifestations of the disease, generally less severe, which may include or even exclusively affect cognitive and/or heart function. Except for this small percentage of female carriers affected by symptoms related to chromosomal rearrangements, disease in most of the cases is caused by an non-random inactivation of the X chromosome.19–21 This document will serve as a guide to improve the quality of care provided to patients diagnosed with DMD, regardless of sex.
MethodsThis document was drafted by a working group including neurologists, paediatric neurologists, and rehabilitators with experience in the management of DMD patients at several Spanish hospitals. We performed a literature review of databases (Medline, PubMed, and Cochrane) using the search strategy: (guidelines [MeSH Terms] OR consensus document [MeSH Terms]) AND (guidelines [MeSH Terms]) AND Duchenne. We limited results to the last 10 years. Google Scholar was also used for Spain. The working group classified and ordered the level of evidence and the grade of recommendation in accordance with the American Academy of Neurology (AAN) criteria (Table 1).22
Criteria for evaluation of the level of evidence and the grade of recommendation according to the AAN.
| Level of evidence | |
|---|---|
| Class I | Randomised controlled clinical trial with the following characteristics: blinded, with a clearly defined primary objective, inclusion and exclusion criteria, and with an appropriate register of drop-outs |
| Class II | Randomised controlled clinical trial not fulfilling one or more of the above mentioned characteristics |
| Class III | Any other controlled trial |
| Class IV | Rest of studies, including consensus documents and expert opinions |
| Grade of recommendation | |
|---|---|
| A | Established as effective, ineffective, or harmful (at least 2 class I studies) |
| B | Probably effective, ineffective, or harmful (a class I study or 2 class II studies) |
| C | Probably effective, ineffective, or harmful (a class II study or 2 class III studies) |
| U | Insufficient or conflicting data |
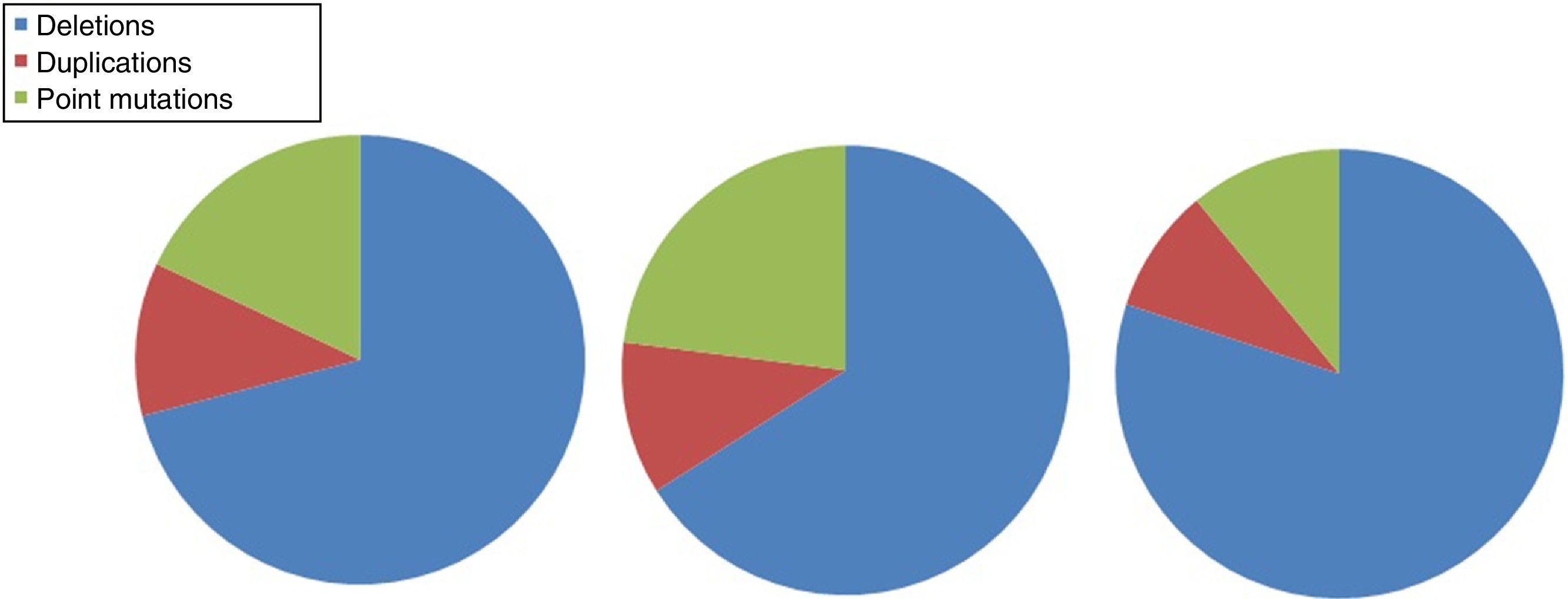
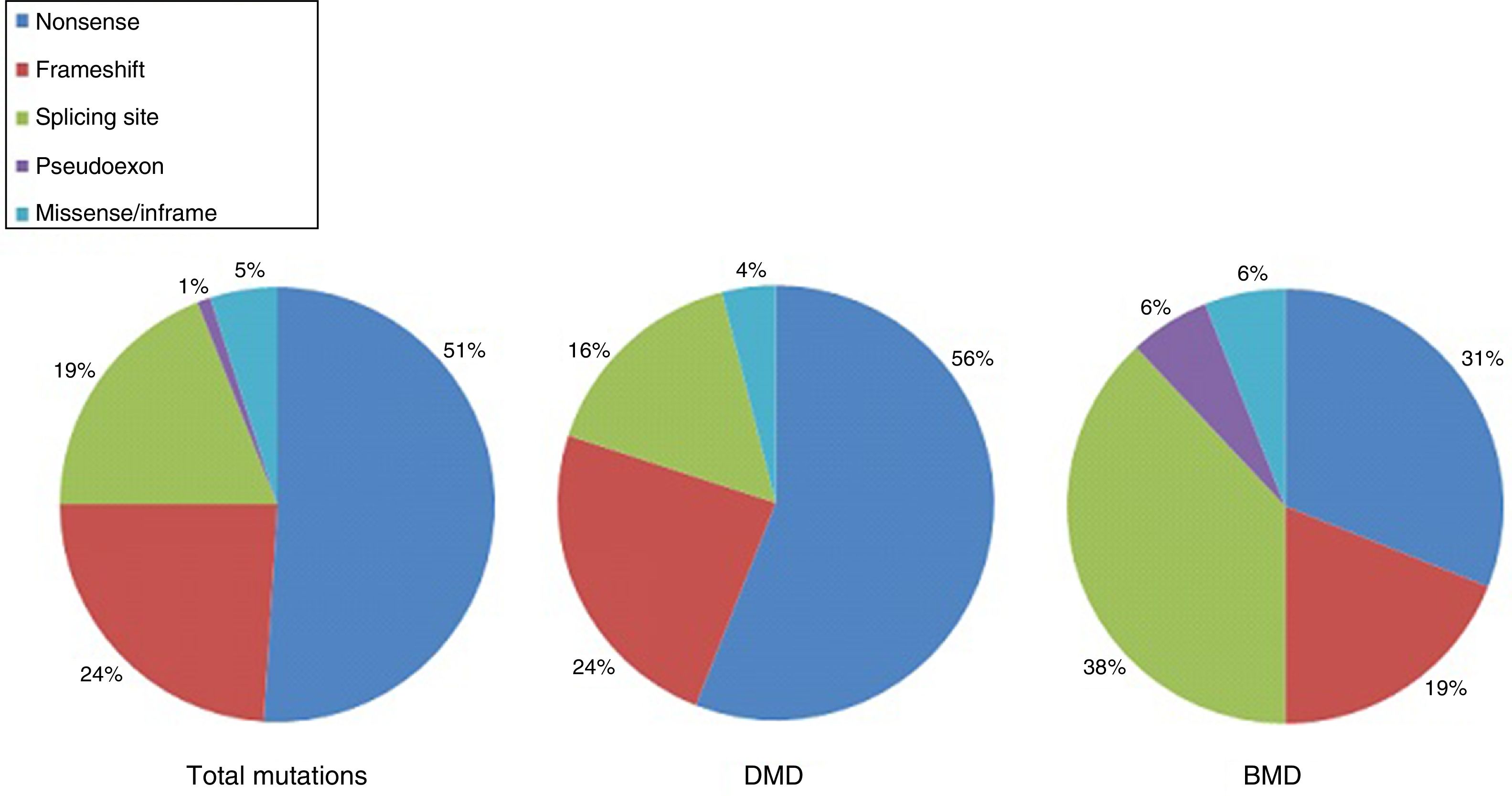
DMD is caused by mutations in the dystrophin gene, located on the X chromosome. Most patients present large gene rearrangements caused by exon deletions (approximately 60%-65%) or duplications (5%-15%). Approximately 20% of cases present small mutations, either point mutations caused by a change in a single nucleotide base or insertion or deletion of one or more nucleotides.23 Of the latter group, nonsense mutations constitute the most frequent type, affecting 10% of all patients.24,25Figs. 1 and 2 show the relative frequency of mutations in a large series of dystrophinopathies in Spanish patients. Regardless of the type of mutation, genetic alterations (with the exception of point mutations) induce a shift in the reading frame of the mRNA that synthesises dystrophin, leading to the production of a non-functional protein.26 However, in 10% of patients this rule does not apply,27,28 as a hidden secondary rearrangement, or spontaneous exon skipping, restores the reading frame. Pathogenic mechanisms due to point mutations are varied; the most frequent consists of nonsense mutations generating a premature stop codon without disrupting the reading frame, causing a non-functional aberrant dystrophin, which also produces a DMD phenotype.29

Distribution of gene mutations associated with DMD and BMD in the Spanish population. Adapted from Juan-Mateu et al.25

Distribution of point mutations associated with DMD and BMD in the Spanish population. Adapted from Juan-Mateu et al.25
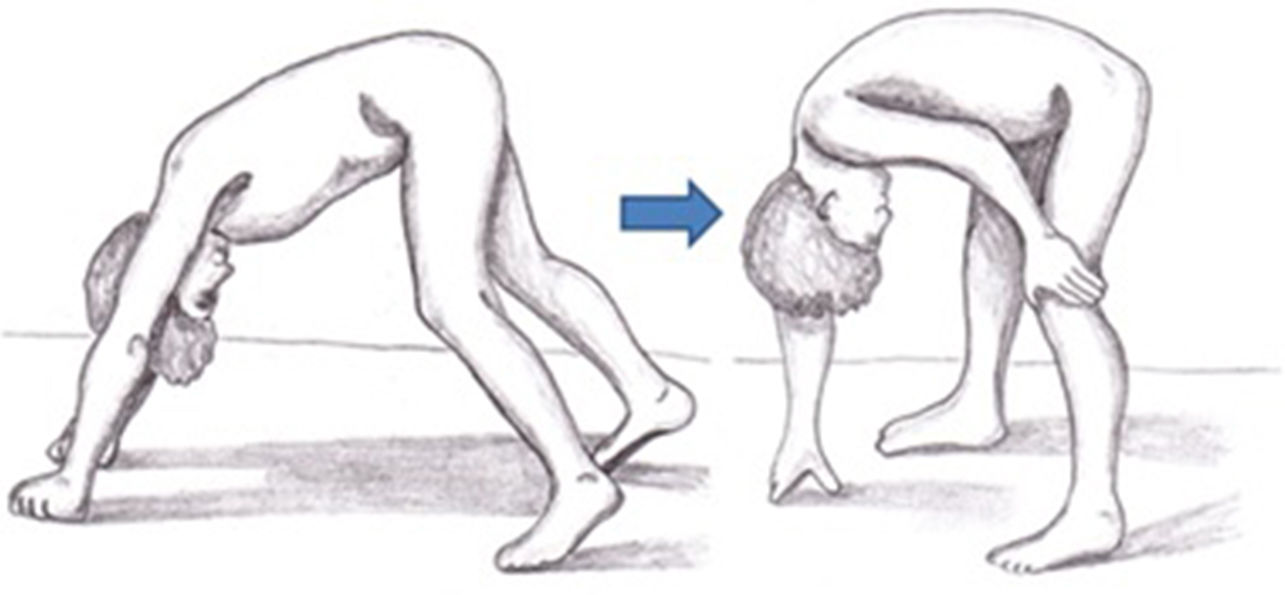
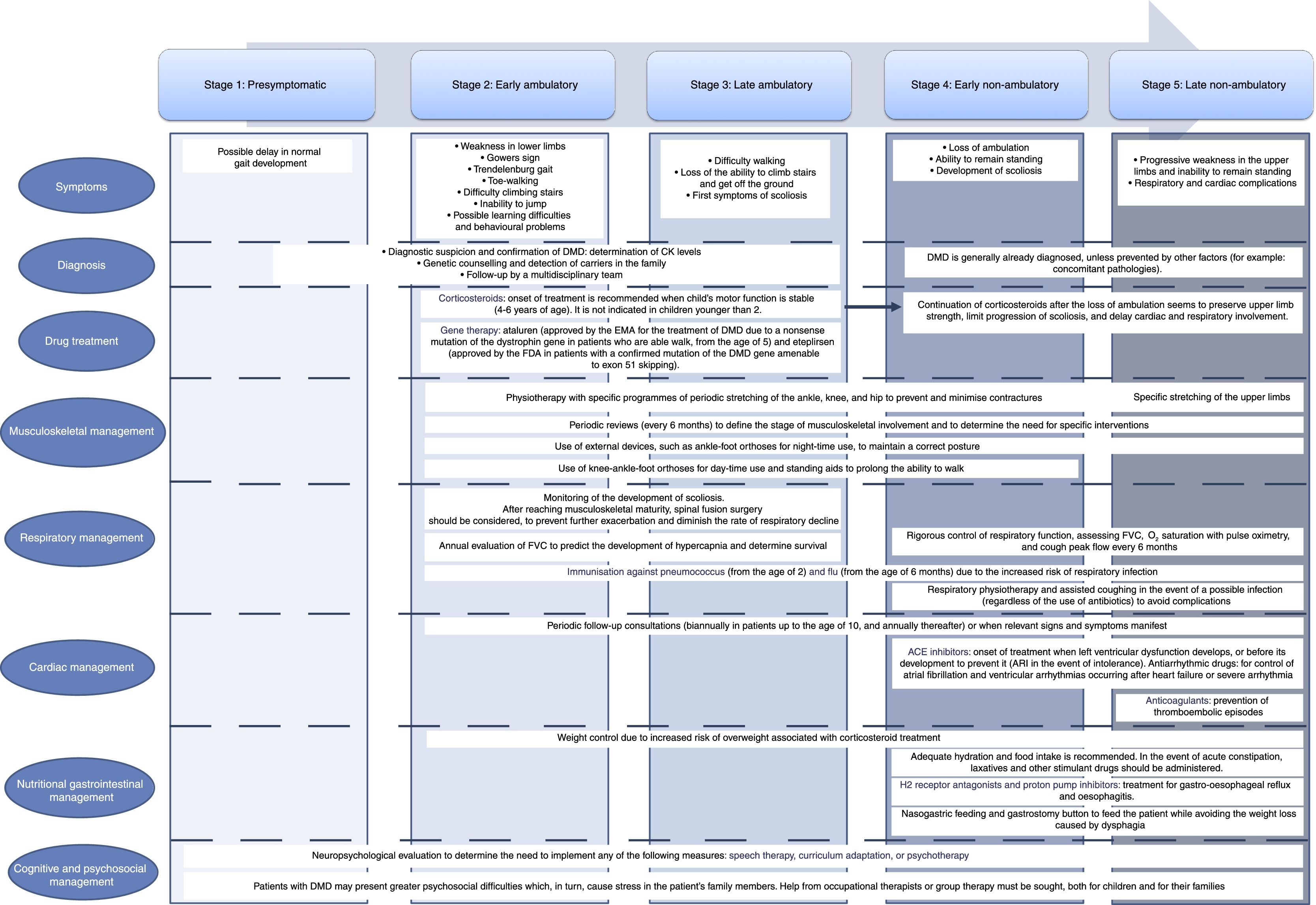
Symptoms of DMD are not visible at birth, but some patients display an early neurodevelopmental delay. Initial symptoms usually manifest during the first 3 years of life as difficulty walking, frequent falls, difficulty climbing stairs and getting off the ground, and a tendency to toe-walking (Table 2).30 Neurological examination performed during this first stage may reveal axial weakness, positive Gowers sign (Fig. 3), muscular pseudohypertrophy, or mild retraction of the Achilles tendon.26 During this stage, cognitive problems begin to manifest in some patients. These children display favourable motor development until 4-6 years of age; development subsequently slows and reaches a plateau between 4 and 8 years,31 then undergoes progressive decline characterised by increasing loss of strength and loss of acquired abilities.8,32 In this phase, children develop contractures and retractions of the least mobile joints,26 which has a negative impact on comfort and motor function.33 Reduced physical activity, together with corticotherapy, promotes the development of overweight, which reduces mobility, favours osteoporosis, and increases the risk of fractures.34 Until recently, patients became reliant on wheelchairs before adolescence.26 However, improvements in support measures and corticotherapy have delayed the loss of the ability to walk.8
Summary of the main clinical characteristics of the different stages of DMD.
| Early ambulatory | Late ambulatory | Early non-ambulatory | Late non-ambulatory |
|---|---|---|---|
| Weakness in the lower limbsGowers signTrendelenburg gaitToe walkingDifficulty climbing stairsInability to jumpLearning difficulties and behavioural problems | Increasing difficulty walkingInability to climb stairs and get off the groundFirst symptoms of scoliosis | Gait lossAbility to standDevelopment of scoliosis | Progressive weakness in the upper limbs and inability to remain seatedCardiac and respiratory complications |

The involvement of dystrophin isoforms that are selectively expressed in other organs, including the brain,26 explains the fact that approximately 20%-34% of patients present intellectual disability, autism spectrum symptoms, or other cognitive or behavioural alterations.35,36
Early and late non-ambulatory phaseIn most cases, patients lose the ability to walk at the age of 12-14 years. This favours the manifestation of severe orthopaedic complications, such as scoliosis due to paraspinal muscle weakness, which affects 90% of the patients; respiratory and cardiac complications are also observed. Respiratory issues manifest during sleep, with nocturnal hypoventilation causing periodic apnoeas, morning headaches, nausea, fatigue, loss of appetite, and cognitive impairment.37,38 Altered respiratory function, aggravated by scoliosis, has long been the main cause of death in these patients. However, proper respiratory care and the different types of ventilatory support now available have led to heart problems (cardiomyopathies and arrhythmias) becoming an important cause of death in these patients.
Table 2 includes the main clinical characteristics of DMD patients throughout the different phases of the disease.
We should point out that the rate of progression and the severity of the disease vary between patients. Significant clinical differences have even been observed between siblings with identical mutations in the dystrophin gene,39 suggesting the presence of environmental or genetic modifying factors which affect the individual phenotype.
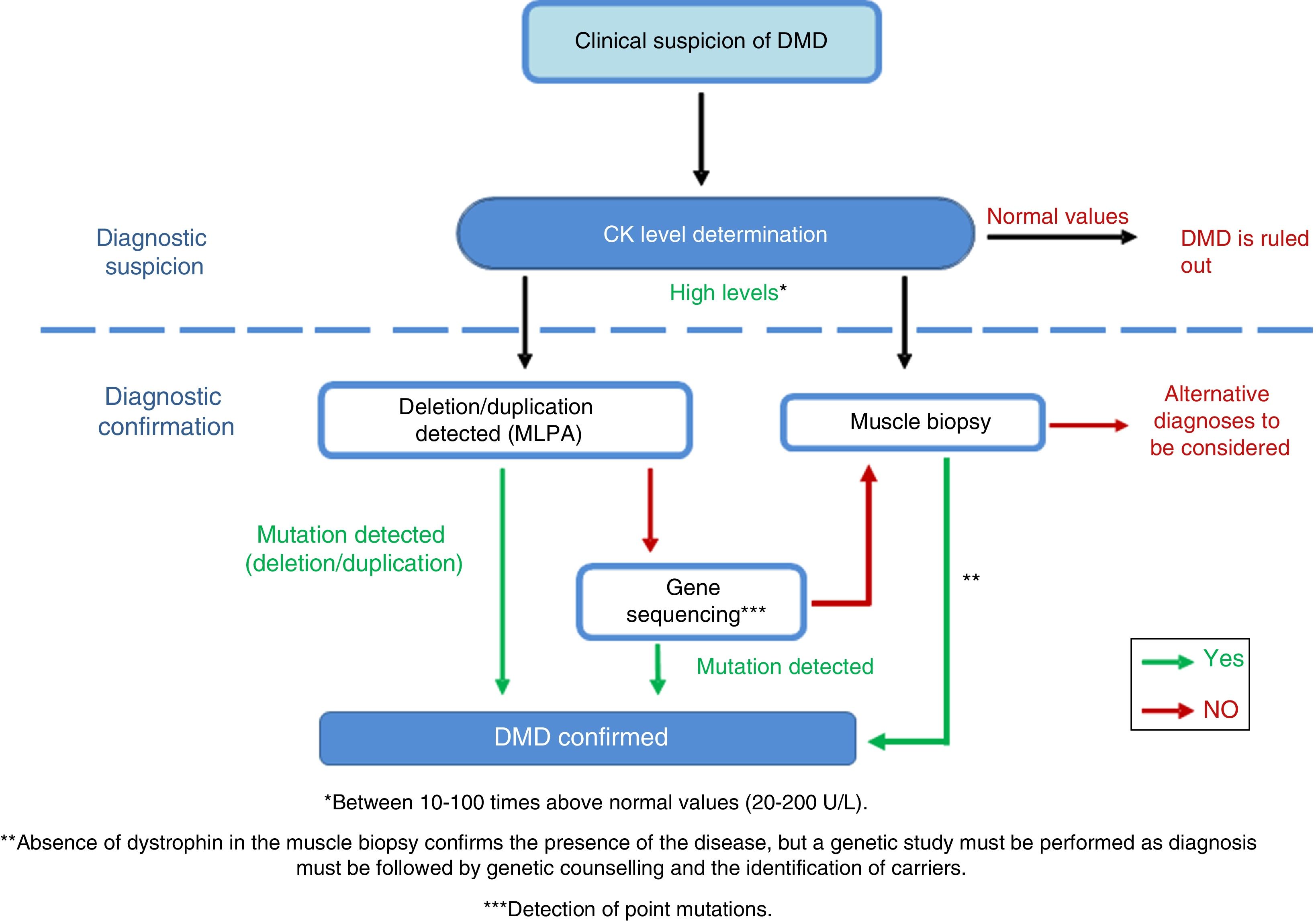
DiagnosisDiagnosis of this pathology must be made as early, as quickly, and as accurately as possible, to ensure early onset of patient intervention. In cases of suspected DMD, serum creatine kinase (CK) levels must be determined (Fig. 4)40 [IV, U]; these are 10-100 times higher26 than normal levels in DMD patients.41 CK levels must be determined in cases of family history of DMD and in the event of the slightest clinical suspicion [IV, U].8,26 Ten percent of female carriers present elevated CK levels.21 Muscle destruction also causes a parallel increase of transaminases, which may lead to erroneous suspicion of a liver disease in presymptomatic children with DMD.

Adaptation of the diagnostic algorithm for DMD, from suspicion to confirmation.
Source: Camacho.26
When DMD is strongly suspected, we recommend performing a genetic study, since this may prevent the need for a muscle biopsy, which is an invasive diagnostic test.26 The first test is usually the multiplex ligation-dependent probe amplification (MLPA) to detect the exons involved in deletions and duplications42 [III, C]. If the test yields positive results and the patient presents a compatible phenotype, a diagnosis of DMD may be established. If results are negative, gene sequencing is performed [III, C] in order to identify point mutations or small deletions/duplications.43,44 The mutation must be fully characterised to assess its influence on the reading frame of the protein, which is the main determinant of the phenotypic diversity of dystrophinopathies.45–47
If the genetic study does not identify any mutation but the patient displays high CK concentrations and signs and symptoms compatible with the disease, a muscle biopsy must be performed to confirm or rule out the diagnosis [IV, U].8,48
The genetic tests described must be complemented with a muscle biopsy. Optical microscopy shows a dystrophic pattern with distortion of the fascicular architecture of the muscle, necrosis, and regeneration of the muscle fibres, as well as increased endomysial adipose connective tissue. Dystrophin deficiency is confirmed by immunohistochemical techniques49 [III, C].
If a muscle biopsy is performed initially and DMD is diagnosed after observation of dystrophin deficiency, genetic tests must be performed to identify the type of mutation causing the disease26 [IV, U]. When biopsy is performed, an investigatory myoblast culture is highly recommended [IV, U].
Treatment and patient follow-upThe progression of DMD has changed in recent years thanks to the implementation of early multidisciplinary treatment including corticosteroid administration and proper respiratory, cardiac, nutritional, physiotherapeutic, and orthopaedic management and follow-up, which stabilises or decreases the rate of progression.
These guidelines include an updated summary of the different approaches to the management and treatment of DMD and its complications, describing both traditional strategies and the most cutting-edge therapies. These are summarised in Appendix 1.

Traditional management is based on the use of corticosteroids, given their long-term beneficial effects on motor, cardiac, and respiratory function (Appendix 1)50 [I, A]. We recommend starting treatment when the child's motor function reaches a stable level (4-6 years of age) [IV, U]. The use of clinical and functional scales may help identify this moment51,52 [III, C]. There is controversy as to whether treatment with corticosteroids should be maintained after loss of ambulation. Recent studies have shown that upper limb strength is preserved, progression of scoliosis is reduced, and cardiac and lung involvement is delayed if corticotherapy is continued53 [IV, U]. The recommended national vaccination programme must be completed before starting treatment with corticosteroids, and immunity to varicella must be demonstrated8,54 [IV, U]. The corticosteroids used are prednisone and deflazacort. Intermittent administration is less effective but is associated with fewer adverse effects.55 The most widely used treatment schedules are: (a) 0.75mg/kg/day prednisone, (b) 0.9mg/kg/day deflazacort, and (c) 0.75mg/kg/day prednisone on alternating days or on 10 consecutive days followed by 10 rest days. Although both drugs have shown a similar efficacy in improving muscle strength and function50 [I, A], more recent studies suggest that continuous treatment with deflazacort is generally safer56 [I, A].
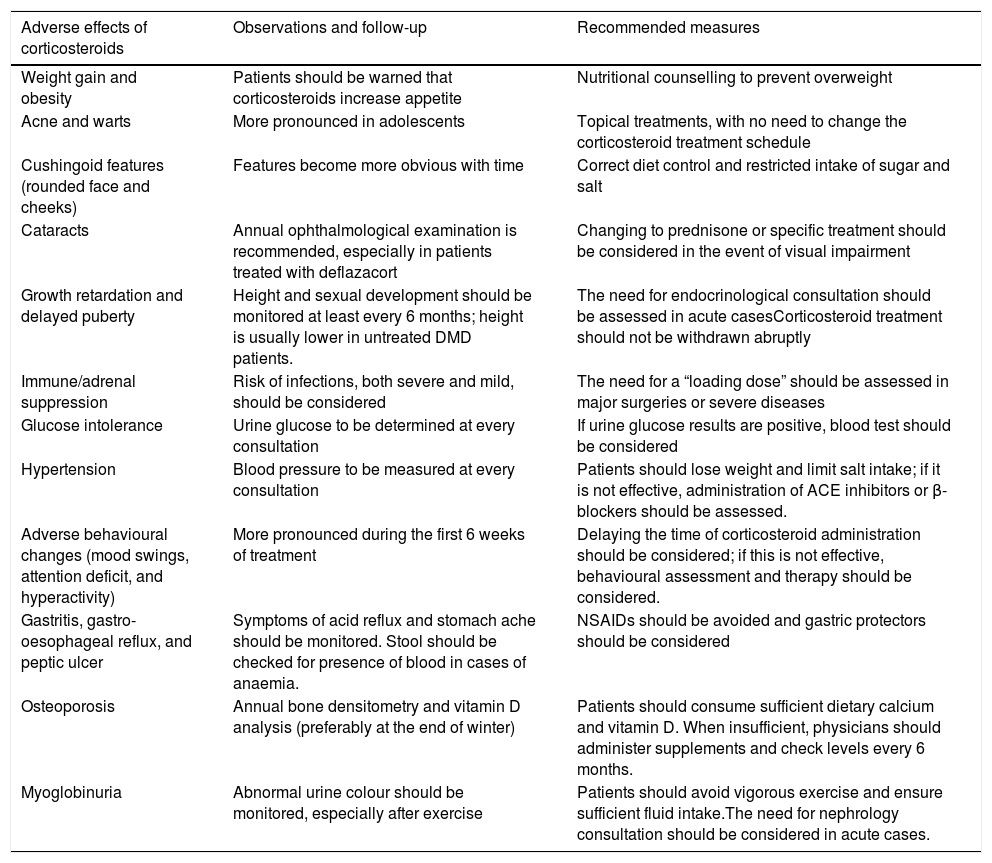
Prednisone and prednisolone have an anti-inflammatory effect and increase skeletal muscle strength,57 whereas deflazacort also promotes the regeneration of these muscles.58,59 A significant percentage of patients do not tolerate the long-term use of corticosteroids and experience adverse effects (Table 3), while others do not respond satisfactorily to treatment.60 Corticosteroid dose is generally increased as the child grows, provided that adverse effects are manageable, until the child weighs approximately 40kg.16 When patients experience no significant adverse effects, the established dose is maintained during the non-ambulatory phase. If adverse effects are not easily manageable, the dose will be reduced by 30% before withdrawal is considered.26
Main adverse effects of corticosteroid treatment; follow-up and recommended therapeutic measures.
| Adverse effects of corticosteroids | Observations and follow-up | Recommended measures |
|---|---|---|
| Weight gain and obesity | Patients should be warned that corticosteroids increase appetite | Nutritional counselling to prevent overweight |
| Acne and warts | More pronounced in adolescents | Topical treatments, with no need to change the corticosteroid treatment schedule |
| Cushingoid features (rounded face and cheeks) | Features become more obvious with time | Correct diet control and restricted intake of sugar and salt |
| Cataracts | Annual ophthalmological examination is recommended, especially in patients treated with deflazacort | Changing to prednisone or specific treatment should be considered in the event of visual impairment |
| Growth retardation and delayed puberty | Height and sexual development should be monitored at least every 6 months; height is usually lower in untreated DMD patients. | The need for endocrinological consultation should be assessed in acute casesCorticosteroid treatment should not be withdrawn abruptly |
| Immune/adrenal suppression | Risk of infections, both severe and mild, should be considered | The need for a “loading dose” should be assessed in major surgeries or severe diseases |
| Glucose intolerance | Urine glucose to be determined at every consultation | If urine glucose results are positive, blood test should be considered |
| Hypertension | Blood pressure to be measured at every consultation | Patients should lose weight and limit salt intake; if it is not effective, administration of ACE inhibitors or β-blockers should be assessed. |
| Adverse behavioural changes (mood swings, attention deficit, and hyperactivity) | More pronounced during the first 6 weeks of treatment | Delaying the time of corticosteroid administration should be considered; if this is not effective, behavioural assessment and therapy should be considered. |
| Gastritis, gastro-oesophageal reflux, and peptic ulcer | Symptoms of acid reflux and stomach ache should be monitored. Stool should be checked for presence of blood in cases of anaemia. | NSAIDs should be avoided and gastric protectors should be considered |
| Osteoporosis | Annual bone densitometry and vitamin D analysis (preferably at the end of winter) | Patients should consume sufficient dietary calcium and vitamin D. When insufficient, physicians should administer supplements and check levels every 6 months. |
| Myoglobinuria | Abnormal urine colour should be monitored, especially after exercise | Patients should avoid vigorous exercise and ensure sufficient fluid intake.The need for nephrology consultation should be considered in acute cases. |
These drugs are the only gene therapy agents to pass preclinical development and reach the clinical trial phase; their efficacy and safety has been demonstrated, leading to approval by regulatory agencies.61
As previously mentioned, 10% of cases of DMD are caused by nonsense mutations resulting in non-functional truncated proteins.
Ataluren favours ribosomal read through of the premature mRNA stop codon present in nonsense mutations, transforming a non-functional truncated protein into a functional protein.62 The drug's efficacy and safety, established by several clinical trials63–65 [II, B, III], led to its conditional authorisation by the EMA in July 2014 for the treatment of DMD caused by a nonsense mutation of the dystrophin gene, in ambulant patients older than 5 years.62
Eteplirsen is an antisense oligonucleotide designed to skip exon 51, enabling the reading frame to be recovered in some specific deletions of the gene's central region (13% of cases of DMD). It has been shown to improve dystrophin levels in a pilot trial [III, C], which led to its approval by the FDA.
Management at the musculoskeletal levelNeuromuscular pathologists, rehabilitators, physiotherapists, and orthopaedic surgeons should jointly promote maintenance of wide-range and symmetrical mobility16 [III, C]. Biannual evaluations are also recommended to identify risk factors and implement relevant measures and recommendations [IV, U]. This evaluation should include such tests as strength measurement using the Medical Research Council (MRC) scale, timed functional tests, and the 6-Minute Walk Test66; also recommended are motor function scales, such as the North Star Ambulatory Assessment in the ambulatory phase67 or the Performance of Upper Limb Scale68 in the non-ambulatory phase [III, C]. The range of motion of joints should also be measured. Furthermore, the use of external devices helps prevent or minimise contractures and/or deformities, as they are designed to maintain correct posture69 [II]. Specifically, ankle-foot orthoses for night-time use are appropriate in all phases of the disease70,71 [III, C], whereas knee-ankle-foot orthoses for day-time use, and even standing aids, are especially useful during the later ambulatory stage and the beginning of the non-ambulatory stage71,72 [IV, U]. Swimming is an excellent activity for these patients. However, they should avoid high-intensity and eccentric exercise26 [IV, U].
Programmes have been developed with the aim of delaying loss of ambulation and preventing complications derived from extended time spent in the seated position. These prolong the ability to walk with lightweight orthoses, and should be implemented during the last stages of ambulation,73 within 3 months from loss of ambulation [III, C]. Severe deformities in club foot must be corrected with Achilles tenotomy, placement of the orthosis at 2-3 days, and gait retraining74 [IV, U], especially in cases in which the degree of retraction interferes with the gait pattern or is asymmetrical. The development of scoliosis should be monitored. Once skeletal maturity is reached, physicians should assess the need for spinal fusion surgery to prevent exacerbation and diminish the rate of respiratory decline75 [III, C]. Occupational therapy is of vital importance in the non-ambulatory phase76 [IV, U].
Respiratory managementGuidelines for correct respiratory management include involving of a pneumologist and a therapist specialised in respiratory physiotherapy77–79 [IV, U]. During the ambulatory phase, forced vital capacity (FVC) should be evaluated annually. After the loss of ambulation, follow-up evaluations should be conducted every 6 months26: FVC while seated, O2 saturation with pulse oximetry, and cough peak flow26 [IV, U]. It is important to conduct sleep studies when FVC values are lower than 60% in order to detect night-time hypoventilation and obstructive sleep apnoea-hypopnoea syndromes80 [III, C]. Early control of these complications is vital to preventing cardiac complications and cognitive impairment81–83 [II, III; C]. Considering the high risk of respiratory infections, patients should be immunised against such pathogens as pneumococcus (in patients older than 2 years) and flu (from 6 months). This is not contraindicated in patients being treated with corticosteroids; however, these patients may present a decreased immune response. In the event of infection, regardless of the use of antibiotics, assisted coughing (respiratory physiotherapy) is important to prevent complications, especially in advanced stages of the disease84 [IV, U].
Cardiac managementCardiac involvement is an increasingly important complication and is becoming one the main causes of death in these patients. It presents as a left ventricular dysfunction, progressing to a dilated cardiomyopathy which causes heart failure and arrhythmias.85,86 Unlike other cardiomyopathies, it usually progresses asymptomatically due to patients’ lack of mobility; therefore, diagnosis and evaluation are based on the relevant tests.85,86 The evaluation should include electrocardiography and echocardiography at diagnosis, followed by biannual assessments until the age of 10 and annual assessments thereafter. Heart MRI scans are being incorporated into the follow-up due to their higher sensitivity in detecting heart dysfunction and fibrosis87,88 [III, C].
Angiotensin-converting enzyme (ACE) inhibitors constitute the first-line treatment.16,85 Recent studies support their use before signs of ventricular dysfunction appear in order to delay onset89,90 [I, A]. In case of intolerance to these drugs, angiotensin receptor inhibitors (ARI) may be used91 [I, B]. Aldosterone receptor antagonists (ARA) have also shown a cardioprotective effect, although further study is needed regarding their use as an alternative to ACE inhibitors or in combined therapy92 [I, U]. The use of β-blockers is more controversial, since there is not sufficient evidence showing a clear benefit for paediatric heart diseases of different aetiologies93 [II, C]; no positive effects were observed when administered in association with an ACE inhibitor92 [II, C].
Treatment with antiarrhythmic drugs to control atrial fibrillation and ventricular arrhythmias is started after heart failure or severe arrhythmia85 [IV, U]. Systemic arterial hypertension and heart rate alterations must be treated8,16 [IV, U].
Gastrointestinal and nutritional managementPatients with DMD present a high risk of weight problems. Periodic nutritional follow-up and adjustments in diet are therefore necessary. At advanced stages of the disease, dysphagia94 may cause weight loss, exacerbating the gradual loss of respiratory muscle strength. In these cases, nasogastric feeding and subsequent placement of a gastrostomy button may be considered. In cases of weight loss with no dietary changes, we must assess the possibility of increased energy expenditure due to respiratory effort, in order to rule out the need to implement non-invasive ventilation (NIV) or adapt NIV parameters16 [IV, U].
Patients in the non-ambulatory phase may experience gastro-oesophageal reflux and oesophagitis due to the involvement of the oesophageal muscles. These symptoms are generally treated with H2 receptor antagonists and proton pump inhibitors, either alone or in combination with prokinetic drugs, sucralfate, and antacids95 [III, C]. Patients are prone to constipation; adequate hydration levels and food intake must therefore be guaranteed. Laxatives and enemas may be used occasionally, as may magnesium hydroxide, lactulose, or polyethylene glycol if symptoms persist.
Cognitive and psychosocial managementDMD predominantly affects the muscles, but may also affect the CNS in several ways, including neurodevelopmental delay in childhood,96 cognitive and learning problems in school-age children,51 and a significantly increased incidence of autism spectrum disorders, attention-deficit/hyperactivity disorder, obsessive-compulsive behaviour, and mood disorders97–99; these may even precede muscular symptoms.100 Generally, incidence of these disorders in different patient samples ranges from 20% to 30%, with symptoms being severe in 5% of cases.96 These disorders should be assessed specifically using such standardised scales as the Griffiths Developmental Scale,101 the Bayley III Scales of Infant and Toddler Development,51 or the Wechsler Scale of Intelligence,35 in addition to other specific questionnaires [III, C]. Depending on the results, speech therapy, psychotherapy, or curriculum adaptation may be indicated26 [IV, U]. For many parents, the stress caused by the child's psychosocial problems and the difficulty of achieving recognition of the disease exceeds the stress related with the physical aspects of the disease; therefore, psychological support may be beneficial for both patients and parents.102
Precautions with anaesthesiaIntravenous anaesthetics are recommended given the high risk of malignant hyperthermia and rhabdomyolysis associated with such inhalation anaesthetics as halothane or isoflurane. Furthermore, depolarizing muscle relaxants are contraindicated26 [IV, U].
Genetic counsellingIdentifying mutation carriers is essential for genetic counselling. It should be noted that one-third of mothers of affected children are not carriers; the risk of transmission is minimal in these cases. A possible germinal mosaicism may give rise to the presence of unexpected mutations in the ova of apparently non-carrier women103; therefore, a prenatal/preimplantation study of all mothers should be performed for a safe prevention.
The importance of genetic counselling in DMD therefore resides partly in identifying possible carriers with the relevant genetic tests. These tests will also provide the necessary clinical information to identify the genetic variant causing the pathology in each case, thereby determining patients’ eligibility for inclusion in clinical trials to assess future therapies for specific mutations.104
Conclusions and implementationDuchenne muscular dystrophy is a severe condition causing early loss of ambulation and the development of complications leading to poorer quality of life and premature death. Although there is currently no curative treatment, there are strategies that delay the natural progression of the disease and the appearance of complications. Precise and early diagnosis is necessary to effectively implement these strategies, as well as the implementation of a predefined, multidisciplinary follow-up plan. Regarding diagnosis, initial recommendations in the event of well-founded suspicion of a DMD case include performing a genetic study to identify possible mutations of the dystrophin gene, which is not invasive and provides an unequivocal result.26 The special interest of the technique resides in the possibility of identifying the specific mutation causing the condition, which will also help direct future therapies specifically at the mutant gene and determine patients’ eligibility for those studies.104
Diagnosis must be completed with the study of carriers. Current management of DMD consists in the use of corticosteroids. Despite some cases of intolerance and refractory symptoms, most corticosteroids have shown significant beneficial effects on heart, lung, and motor function,105 even after the loss of ambulation.53 To this fundamental therapy, we should add respiratory, cardiac, physiotherapeutic, orthopaedic, gastrointestinal, and nutritional measures aimed at reducing symptoms and improving patients’ quality of life. These should be implemented by a multidisciplinary team specialising in the management of DMD.8 Advances in respiratory management, especially the use of nocturnal mechanical ventilation, have significantly increased survival in DMD.79,106
Treatment of the cardiomyopathy associated with DMD should be started before signs of abnormal heart function appear,8 in order to ensure improvements in the progression of the heart disease.92 Regarding musculoskeletal management, it is essential to monitor scoliosis progression and promote rehabilitation. Therefore, a multidisciplinary approach is the cornerstone of optimal care to DMD patients. In conclusion, it is important to bear in mind that, despite the important advances achieved with corticosteroid treatment in increasing patients’ life expectancy, as well as the latest innovations in gene therapy (such as ataluren and eterlipsen), research into DMD should continue to provide new concepts and strategies, constantly improving the management of the disease.
FundingEditorial support was provided by Patricia Ortega, from the medical department at Adkhoma Health Research, S.L. The study was financed by PTC Therapeutics S.L. through an independent research grant. The study design, text, and opinions expressed in this work are the exclusive responsibility of the authors.
Authors’ contributionsAll authors contributed their expertise in the conception, design, and performance of this study, as well as in the literature search and interpretation of data, and participated in the drafting and review of the article. All authors approve the final version.
Conflicts of interestAna Camacho, Julita Medina, Juan Jesús Vilchez Padilla, Andrés Nascimento, and Marcos Madruga have worked as consultants for PTC Therapeutics and have received fees for giving training talks. The other authors have no conflicts of interest to declare.
Please cite this article as: Nascimento Osorio A, Medina Cantillo J, Camacho Salas A, Madruga Garrido M, Vilchez Padilla JJ. Consenso para el diagnóstico, tratamiento y seguimiento del paciente con distrofia muscular de Duchenne. Neurología. 2019;34:469–481.
