












Spinal muscular atrophy (SMA) is a neurodegenerative disease caused by a biallelic mutation of the SMN1 gene, located on the long arm of chromosome 5, and predominantly affects the motor neurons of the anterior horn of the spinal cord, causing progressive muscle weakness and atrophy. The development of disease-modifying treatments is significantly changing the natural history of SMA, but uncertainty remains about which patients can benefit from these treatments and how that benefit should be measured.
MethodologyA group of experts specialised in neurology, neuropediatrics, and rehabilitation and representatives of the Spanish association of patients with SMA followed the Delphi method to reach a consensus on 5 issues related to the use of these new treatments: general aspects, treatment objectives, outcome assessment tools, requirements of the treating centres, and regulation of their use. Consensus was considered to be achieved when a response received at least 80% of votes.
ResultsTreatment protocols are useful for regulating the use of high-impact medications and should guide treatment, but should be updated regularly to take into account the most recent evidence available, and their implementation should be assessed on an individual basis. Age, baseline functional status, and, in the case of children, the type of SMA and the number of copies of SMN2 are characteristics that should be considered when establishing therapeutic objectives, assessment tools, and the use of such treatments. The cost-effectiveness of these treatments in paediatric patients is mainly influenced by early treatment onset; therefore, the implementation of neonatal screening is recommended.
ConclusionsThe RET-AME consensus recommendations provide a frame of reference for the appropriate use of disease-modifying treatments in patients with SMA.
La atrofia muscular espinal (AME) es una enfermedad neurodegenerativa, causada por una mutación bialélica del gen 5q SMN1, que afecta predominantemente a las neuronas motoras de la asta anterior medular causando una progresiva debilidad y atrofia muscular. La aparición de tratamientos modificadores del curso de la enfermedad está cambiando considerablemente la historia natural de la AME, pero existe todavía incertidumbre sobre qué pacientes se pueden beneficiar de estos tratamientos y cómo se debería medir ese beneficio.
MetodologíaUn grupo de expertos especialistas en neurología, neuropediatría, rehabilitación y de la asociación de pacientes FundAME analizaron, siguiendo la metodología Delphi, 5 apartados relacionados con el uso de los nuevos tratamientos: aspectos generales; objetivos del tratamiento; herramientas de medición de resultados; requisitos de los centros tratantes; y regulación de su uso. Se definió como consenso cuando una respuesta recibió al menos el 80% de los votos.
ResultadosLos protocolos de tratamiento son útiles para regular el uso de medicamentos de alto impacto y deben constituir una guía para el tratamiento, pero se deben actualizar regularmente para recoger la evidencia más reciente disponible y su implementación se debe valorar de forma individualizada. La edad, la funcionalidad basal y, en el caso de los niños, el tipo de AME y el número de copias de SMN2 son características que se deben tener en cuenta a la hora de establecer los objetivos terapéuticos, las herramientas de medición y el uso de dichos tratamientos. El aspecto más determinante del coste-efectividad de estos tratamientos en la edad pediátrica es su inicio precoz, por lo que se recomienda la instauración de un cribado neonatal.
ConclusionesLas recomendaciones del consenso RET-AME proporcionan un marco de referencia para el uso adecuado de tratamientos modificadores de la enfermedad en pacientes con AME.
Spinal muscular atrophy (SMA) is an autosomal recessive neurodegenerative disease caused by a biallelic mutation of the SMN1 gene, located on chromosome 5q. The mutation results in a deficiency of the survival of motor neuron (SMN) protein and progressive degeneration of alpha motor neurons of the ventral horn of the spinal cord, causing progressive muscle weakness and atrophy.1
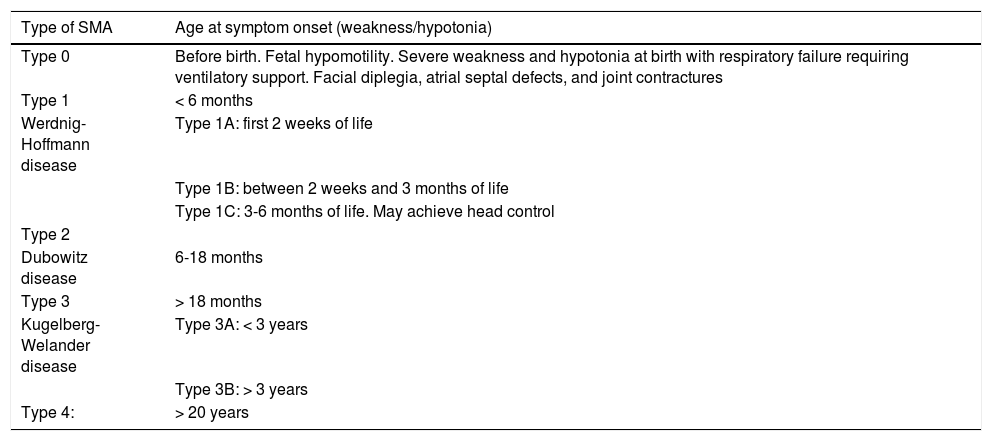
SMA has traditionally been classified into 5 broad categories according to the maximum motor developmental milestone achieved and the age of clinical onset (Table 1); this enables us to establish a working prognosis in untreated patients. Given the progressive nature of the disease, and according to progression time and the treatments administered, the established type of SMA may not be representative of a given patient’s current functional status. Therefore, it is also useful to classify patients according to their baseline functional status, as walkers (can independently walk at least a few steps), sitters (cannot walk but can sit unsupported), and non-sitters (cannot sit independently).1,2
Traditional classification of spinal muscular atrophy.
| Type of SMA | Age at symptom onset (weakness/hypotonia) |
|---|---|
| Type 0 | Before birth. Fetal hypomotility. Severe weakness and hypotonia at birth with respiratory failure requiring ventilatory support. Facial diplegia, atrial septal defects, and joint contractures |
| Type 1 | < 6 months |
| Werdnig-Hoffmann disease | Type 1A: first 2 weeks of life |
| Type 1B: between 2 weeks and 3 months of life | |
| Type 1C: 3-6 months of life. May achieve head control | |
| Type 2 | |
| Dubowitz disease | 6-18 months |
| Type 3 | > 18 months |
| Kugelberg-Welander disease | Type 3A: < 3 years |
| Type 3B: > 3 years | |
| Type 4: | > 20 years |
SMA: spinal muscular atrophy.
Several approved treatments are currently available for SMA that increase levels of the SMN protein, either through replacement of SMN1 (onasemnogene abeparvovec) or by promoting the inclusion of exon 7 in the transcription of SMN2 (nusinersen and risdiplam). Through these mechanisms, these drugs reduce neuronal death and muscle atrophy.3
The emergence of these treatments is changing the natural history of the disease, giving rise to new phenotypes. For example, the concept of children with presymptomatic SMA has recently been introduced.4 While there is currently no consensus on this subject, presymptomatic SMA may be defined as normal motor development, presence of tendon reflexes, ulnar compound motor action potential (CMAP) amplitude greater than 1-1.5 mV, and absence of the following signs and symptoms5,6: weakness or hypotonia, tongue fasciculations, diaphragmatic paradoxical breathing, bell-shaped thorax, hypoxaemia, hypercapnia, and swallowing and feeding problems.
Nusinersen, the first approved treatment for SMA, was marketed in Spain in 2018 under a pricing and reimbursement system that includes a prescribing protocol and a register of treatment effectiveness.7 This enables rapid, equitable, and reasonably generalised access to treatment, and guarantees that results are evaluated in the medium and long term.
However, since the approval of the protocol, significant advances have been made in clinical experience and in the information available on the natural history of the disease, tools for evaluating outcomes in different phenotypes, and the real-world efficacy of the different treatments and the factors influencing efficacy, among other areas. Our working group was created to establish a series of consensus recommendations that may serve as a basis for a new prescribing protocol and to promote proper use of the new drugs.
Material and methodsDelphi methodThe Delphi method is an information-gathering technique enabling consensus to be established by a group of experts in a given field through repeated consultations.8 It is particularly useful for establishing consensus in groups comprising 5-30 members, to address complex problems, and in situations of uncertainty or when insufficient objective information or scientific evidence is available.8 In these cases, it is appropriate to use expert judgement, whose reliability is strengthened as the limitations of a single individual are overcome by establishing a collective judgement.8 The main characteristics of this methodology are the following: 1) in an iterative process, participants may express their opinions on several occasions and are offered the opportunity to reflect on these with reference to the majority opinion; 2) anonymity is ensured, decreasing the risk of prestige or leadership bias; 3) development of the process is controlled by the Delphi coordinators; and 4) though the information gathered is qualitative, it may be structured in such a way that the results are representative of the opinions of the working group.
Study designThe study was conducted in 2 phases. In a first phase, in June 2020, a group of physiatrists (E. Ibáñez, M. Martínez-Moreno, J. Medina), neurologists (M. Povedano, J.F. Vázquez-Costa), and paediatric neurologists (D. Gómez-Andrés, M. Madruga, F. Munell, A. Nascimento, S.I. Pascual, I. Pitarch) contacted other Spanish specialists with experience and interest in the treatment of patients with SMA, seeking the most representative possible geographic distribution. We also contacted FundAME, a patients’ association. Thus, we created a panel of 23 specialist physicians, including one representing FundAME. Each member of the panel was asked to submit a free-form text analysing the current treatment protocol for SMA,7 including the aspects they considered should be included and/or updated in a future protocol. Based on the responses, it was agreed that the following areas would be addressed: 1) general considerations; 2) treatment objectives; 3) evaluation of outcomes; 4) requirements for treatment centres; and 5) regulation of the use of the new treatments (including criteria for indication, ineligibility, and suspension).
In the second phase, between July 2020 and February 2021, and in accordance with the Delphi methodology,8 the study coordinators (IP and JFVC) prepared questionnaires, having previously evaluated the ideas and suggestions proposed by the participants. Given the panel’s diversity in terms of training and experience, and marked differences in the populations analysed, different questionnaires were designed for adult and paediatric patients for most areas. IP was responsible for coordinating the consensus for paediatric patients, and JFVC coordinated the consensus process for adult patients. While all questionnaires were open to all participants, we asked each panel member only to respond to questionnaires addressing subjects in which he/she had experience. A minimum of 10 responses were required for the conclusions drawn from each questionnaire to be considered valid. Questionnaires were distributed to members of the expert panel approximately every 2 weeks, using an online platform ensuring responses were anonymous. Consensus was considered to be very strong when more than 80% of participants voted for a particular response. For the remaining questions, some responses were reformulated to facilitate consensus; for example, responses receiving fewer than 10% of votes were eliminated, or new suggestions from the expert panel were added. These questions, along with statistics on participants’ responses from the first round, were distributed again for reconsideration by the expert panel. After this second round, participants were invited to debate questions for which consensus had not been reached, before questions were again submitted for a third round. After the third round, we considered consensus to have been reached for responses on which more than 80% of participants were in agreement, and reduced consensus for responses receiving 70%-–80% of votes. When a response received fewer than 70% of votes, we considered consensus not to have been reached, and excluded that question from the recommendations. Therefore, the results only include responses for which strong or very strong consensus was reached.
Finally, a draft of the consensus positions reached was forwarded to the panel members after completion of each section, and a teleconference was held in April 2021 to analyse the results and draw conclusions.
Due to restrictions associated with the COVID-19 pandemic, the entire process was conducted remotely.
No pharmaceutical company had any direct or indirect involvement in the consensus process, the preparation of the recommendations, or the drafting of the article.
ResultsGeneral considerationsFirstly, we analysed what subjects should be addressed, and in what way, in a protocol regulating the use of new treatments for SMA. The consensus recommendations are shown in Table 2. To summarise, the protocol should constitute a set of treatment guidelines, but its implementation should be assessed on an individual basis. Thus, such characteristics as patient age, baseline functional status, and (in paediatric patients) type of SMA and the number of SMN2 copies should be taken into account when establishing treatment objectives, selecting assessment tools, and indicating the new treatments.
Consensus recommendations on a protocol regulating the use of the new treatments for spinal muscular atrophy.
| A) The protocol must establish a set of guidelines for the treatment and follow-up of patients with SMA, although their implementation must be considered on an individualised basis for each patient and context in order to ensure rational use of the new drugs and the greatest possible benefit to the patient. |
| B) Any protocol for follow-up and regulation of disease-modifying treatments for SMA should address the following issues: |
| - Treatment objectives |
| - Eligibility criteria |
| - Patient follow-up |
| - Criteria for suspension |
| - Requirements for treatment centres |
| - Therapeutic algorithm (what drugs to use in which patients). |
| C) Such protocols should be revised every 2-3 years, unless there are substantial changes in the treatments or the evidence available. |
| D) Given the heterogeneity of the disease, all protocols assessing and regulating the use of disease-modifying drugs should differentiate between patient subgroups, according to baseline characteristics. When establishing subgroups, it is essential to consider the characteristics of the patient, specifically: |
| - Age: adult (> 15 years) or paediatric patients (≤ 15 years). Paediatric patients should be classified as infants (< 2 years), preschool children (2-6 years), schoolchildren (7-10 years), and prepubescents (11-15 years). |
| - Baseline functional status (walkers, sitters, or non-sitters) |
| - In paediatric patients, type of SMA (presymptomatic or symptomatic) and number of SMN2 copies must also be considered. |
| E) National and/or regional mechanisms (expert committees) are needed to evaluate and resolve cases in which the clinical judgement of the treating physician diverges from the recommendations of the treatment protocol. |
| F) SMA should be included in neonatal screening tests, as prognosis depends on the time of treatment onset. |
SMA: spinal muscular atrophy.
Consensus positions on treatment objectives are shown in Table 3. In brief terms, we should consider 2 main objectives: efficacy and long-term safety. Where possible, these should be assessed on an individual basis before treatment is started in each patient. Efficacy may be evaluated in several clinical areas; while the general objective should be to achieve improvements in one or several of these, stabilisation of some areas may also be a valid objective in patients whose history would lead us to expect functional worsening.
Consensus treatment objectives.
| A) Two broad objectives should be considered: efficacy and long-term safety. Where possible, these should be assessed on an individual basis before starting treatment. |
| B) The treatment objectives set for each patient should take into account at least the following variables: |
| 1) Baseline functional status (walkers, sitters, or non-sitters) |
| 2) Type of SMA |
| 3) Current age |
| 4) Disease course/phase. As the progression of SMA is not linear (especially in childhood), it is essential to know the previous course of the disease and the current phase of the disease in each patient at all times. In paediatric patients, the classification for which we found greatest consensus (reduced consensus: 77.8%) includes 4 phases: |
| - Asymptomatic phase |
| - Symptom onset phase (onset) |
| - Chronic phase (stability) |
| - Decline phase (decline) |
| 5) Presence of scoliosis, history of scoliosis surgery, or contractures. |
| C) For each patient, several objectives should be set in different therapeutic areas (motor, respiratory, and bulbar function; quality of life). |
| D) The following efficacy variables (listed in order of strength of consensus) should be evaluated in all patients: |
| 1) Motor function: improved motor function should be the objective in the majority of patients, although motor stabilisation may be a treatment objective in certain patients or during certain disease phases. |
| 2) Quality of life: improved quality of life should be a treatment objective in all patients. |
| 3) Respiratory function: improvement or stabilisation of respiratory function should be a treatment objective in all patients. Furthermore, in patients with poor expected survival (SMA types 1 and 2A), survival without ventilatory support should be a treatment objective. |
| 4) Bulbar function (speech and swallowing): improvement or stabilisation of bulbar function may be a treatment objective in affected patients (reduced consensus: 77.8%). |
| 5) Reduction of hospital admissions (reduced consensus: 77.8%) |
| 6) Fatigue and fatigability: improvement should be a treatment objective in affected patients (reduced consensus: 72.2%). |
| E) Stabilisation of certain areas (motor, respiratory, or bulbar function; quality of life) may be considered a treatment objective, as long as improvement in other areas is established as an objective. |
SMA: spinal muscular atrophy.
In order to determine the efficacy of treatment, there is a need for tools to evaluate outcomes (hereinafter, tools) that are appropriate for the characteristics of each subject, and proper training to administer these tools. The panel agreed that at least one tool should be used to measure functional status (one scale for motor function and another multidimensional tool), one for patient-reported outcomes, one to assess respiratory function, and another to evaluate bulbar function. General recommendations for evaluating outcomes are summarised in Table 4, and the tools selected in the consensus process are listed in Table 5 (paediatric patients) and Table 6 (adults).
Characteristics of tools for evaluating outcomes.
| A) New tools should be developed and validated to evaluate the efficacy of new treatments, as the existing tools present limitations, particularly in patients with poorer functional status and in adult patients. |
| B) Tools should measure both qualitative and quantitative improvements: |
| - Quantitative improvements: improvements in motor function (strength, mobility, independence, fatigability, increased CMAP), respiratory function, and bulbar function |
| - Qualitative improvements: quality of life, impact on daily life. |
| C) Raters should be adequately trained to apply each scale or test. |
| D) The scales and tests administered to evaluate treatment response should be adapted to the objectives set for each patient, taking into account their individual characteristics (functional status, type of SMA, age, comorbidities, and progression time/phase). |
| E) Consultations for the assessment of treatment response should not last longer than 90 minutes (including application of scales). |
| F) The interval between consultations to assess treatment response should be established on an individual basis in accordance with patient characteristics and treatment response, with intervals of 4-12 months as a reference. |
| G) Ideally, the conditions of assessment consultations should be standardised (same time, same rater, etc). |
CMAP: compound motor action potential; SMA: spinal muscular atrophy.
Tools for evaluating outcomes in paediatric patients.
| Motor function/functional status: |
| A) Presymptomatic patients diagnosed by screening tests should be assessed with at least one motor function scale for SMA (CHOP INTEND) and according to the WHO41 or HINE2 motor developmental milestones.42 The objective of follow-up in untreated presymptomatic patients is to detect the onset of impairment as early as possible (before clear clinical manifestation). In this regard, we established a consensus on testing CMAP, with the addition of other biomarkers (eg, neurofilament) when sufficient evidence is available, as well as some motor scale. |
| B) Motor function should be measured in consultations with paediatric patients, with application of several motor scales, according to the patient’s age and functional status: |
| • < 24 months: CHOP INTEND43,44 |
| • > 24 months (walkers): HFMSE45–48 + 6MWT49–51 |
| • > 30 months (non-walkers): HFMSE45–48 + RULM48 |
| • Functional status should be assessed with the EK2 scale29,30 in non-walkers older than 4 years. |
| Patient-reported outcomes: |
| C) Pediatric Quality of Life Inventory52 (neuromuscular module), administered by caregivers of patients aged 2-18 years and by patients aged 5-18 years. Reduced consensus (78%) |
| Respiratory function: |
| D) We recommend performing spirometry studies including FVC, FEV1, and the FVC/FEV1 ratio (%) from the age of 6 years, although this may be attempted from 4-5 years in collaborative patients. |
| E) Where required, pulmonary function should be assessed by capnography53 in infants. |
| Bulbar function: |
| F) Bulbar symptoms (dysarthria, dysphagia, sialorrhea, etc) should be assessed routinely in children,26,27 although there was no consensus on the specific tools to be used.28 |
6MWT: Six-Minute Walk Test; CHOP INTEND: Children’s Hospital of Philadelphia Infant Test of Neuromuscular Disorders; CMAP: compound motor action potential; EK2: Egen Klassifikation Scale Version 2; FEV1: forced expiratory volume in 1 second; FVC: forced vital capacity; HINE2: Hammersmith Infant Neurological Examination 2; HFMSE: Hammersmith Functional Motor Scale Expanded; RULM: Revised Upper Limb Module; SMA: spinal muscular atrophy; WHO: World Health Organization.
Tools for evaluating outcomes in adult patients.
| Motor function/functional status: |
| A) In walkers, we recommend using the 6MWT49–51 and the ALSFRS-R functional scale.21,22 Alternatively, or additionally, the HFMSE45–48 may be used (reduced consensus). |
| B) In sitters, we recommend using the RULM motor scale48 and the EK2 functional scale.29 Alternatively, or additionally, the HFMSE45–48 may be used (reduced consensus). |
| C) In non-sitters, we recommend using the RULM motor scale48 and the EK2 functional scale.29,30 |
| Patient-reported outcomes: |
| D) We established consensus that the Global Impression of Change54 should be administered to all patients (walkers, sitters, and non-sitters); the scale, completed by the physician or the patient/their caregiver at each consultation, rates the patient’s global health status on a 7-point ordinal scale. |
| Respiratory function: |
| E) In walkers, spirometry testing including FVC should be performed. Alternatively, or additionally, MEP may be measured. |
| F) In sitters, spirometry testing including FVC should be performed. Alternatively, or additionally, MEP may be measured or the daily number of hours using non-invasive ventilation may be recorded. |
| G) In non-sitters, the number of hospital admissions due to respiratory infections should be recorded. Alternatively, or additionally, spirometry testing including FVC may be used or the daily number of hours using non-invasive ventilation may be recorded. |
| Bulbar function: |
| H) No consensus was established regarding how to assess bulbar function in sitters and non-sitters. The most popular option (55.6%) was to use the EK2 items evaluating bulbar function.29,30 |
6MWT: Six-Minute Walk Test; ALSFRS-R: Revised Amyotrophic Lateral Sclerosis Functional Rating Scale; EK2: Egen Klassifikation Scale Version 2; FVC: forced vital capacity; HFMSE: Hammersmith Functional Motor Scale Expanded; MEP: maximum expiratory pressure; RULM: Revised Upper Limb Module.
The panel agreed that the treatment and follow-up of patients with SMA should take place at centres able to provide multidisciplinary care, with specialists trained in the management of neuromuscular disorders. We recommend a minimum accredited level of experience (follow-up of at least 5 patients with SMA) and training at centres treating these patients and assessing treatment response.
Regulation of the new drugsThe general recommendations for indication, ineligibility, and suspension of the new treatments are presented in Tables 7–10.
General recommendations.
| A) Treatment decisions should be made by multidisciplinary teams. |
| B) Prior to the indication of disease-modifying therapy, patients and/or their parents/guardians should be informed of the available options, the risk-benefit balance of each drug, and general conditions of follow-up (criteria for indication and suspension of the treatment). |
| C) Treatment decisions must be made jointly with patients and/or their parents/guardians, whose informed consent must be obtained. |
Criteria for indication.
| General requirements: |
| A) Genetic study of the biallelic mutation of the SMN1 gene |
| B) Patient is receiving care that meets and is expected to continue to meet the guidelines of the Declaración de consenso de normas para el cuidado de la atrofia muscular espinal (“Consensus statement on regulations for the management of spinal muscular atrophy”), including vaccines, prophylaxis, nutritional and respiratory support, or physiotherapy. |
| C) Treatment must plausibly be efficacious for the patient in question. Potential efficacy should be assessed by the physician responsible for treatment, taking into account the drug’s action mechanism, patient characteristics, and the best available evidence at the time. |
| Paediatric patients: |
| A) It is essential to implement neonatal screening for SMA in order to start treatment during the asymptomatic phase. |
| B) Where indicated, treatment should be started as early as possible. |
| C) Presymptomatic children: |
| - Presence of 1-3 copies of the SMN2 gene |
| - Absence of symptoms at the time of treatment onset. |
| D) Patients with type 0 and type 1A SMA: |
| - No consensus was established on disease-modifying therapy in these patients; therefore, individualised assessment by an expert committee is recommended. |
| E) Symptomatic patients (types 1B to 3): |
| - Functional impairment observed in motor and/or functional scales. |
| Adult patients: |
| F) Types 1 to 4: |
| - Functional impairment observed in motor and/or functional scales |
| - Deterioration of functional status defined by medical history interview or assessments conducted over the previous 5 years. |
SMA: spinal muscular atrophy.
Ineligibility criteria.
| A) Unfavourable result of the risk-benefit analysis by the responsible physician, based on the best available evidence at the time |
| B) Very advanced clinical situations with minimal functional activity and need for support in all activities of daily living, which according to clinical judgement are not reversible and cannot be expected to benefit substantially from the drug |
| C) Patients requiring permanent ventilation (typically defined as > 16 hours per day), not caused by an intercurrent acute process, and considered to be irreversible |
| D) Off-label administration, unless justified by emerging evidence |
Criteria for suspending treatment.
| General requirements: |
| A) Continuity of treatment depends on whether the treatment objectives established for each patient are met. |
| B) The expected natural history of the disease in each patient should be taken into account when evaluating whether to suspend treatment. |
| C) Generally, suspension of treatment should be considered in the event of any of the following circumstances: |
| - The patient shows no improvement with respect to the expected natural history of the disease. |
| - The clinician considers the treatment to have insufficient benefit. |
| - The patient’s clinical status worsens due to a clinical situation that is difficult to reverse (eg, worsening respiratory function requiring permanent ventilatory support, not due to an acute episode). |
| - The patient presents severe adverse drug reactions. |
| D) If a treatment is suspended, indication of another of the approved drugs should be considered. |
| E) If a patient worsens after a treatment is suspended, it may be indicated again if deemed appropriate by the responsible clinician. |
| Paediatric patients: |
| A) Treatment should be suspended after sustained, consistent worsening of scores on the motor scale selected for follow-up and/or loss of an acquired motor developmental milestone. |
| - Patients with type 1B/1C SMA: suspension of the treatment should be considered every 12 months (reduced consensus: 75%). |
| - Patients with type 2/3 SMA: suspension of treatment should be considered every 2 years. |
| Adult patients: |
| A) Suspension of treatment should be considered every 2 years (reduced consensus: 71%). After this period, suspension should be considered if: |
| - At least 2 of the treatment objectives established for the patient are not met, including at least one objective measured by the rater (motor scales, respiratory studies, etc) and one reported by the patient (quality of life, functional impact, etc). |
| - The patient presents worsening in one or more treatment objectives, without achieving improvements in other areas. |
| - If patients present improvements for some objectives and worsening for others, decisions must be made on an individual basis, taking into account the objectives, patient characteristics, and the natural history of the disease. |
SMA: spinal muscular atrophy.
It is important to be aware that disease-modifying therapies for SMA should be started as early as possible in paediatric patients, except in those presenting clear symptoms in the first 2 weeks of life; in this period, decisions must be made on an individual basis, with the support of expert committees, if needed. Regarding this point, it is essential to implement neonatal screening for SMA and to start treatment in the presymptomatic stage in children with 1-3 copies of SMN2.
Other relevant factors in the use of the new treatmentsThe following factors should be taken into account in decisions on the indication, exclusion, or suspension of treatment:
- a
Minimal baseline functional status
- b
Severe scoliosis and contractures
- c
Limited data from trials and clinical practice
- d
Difficulty of access, in the case of intrathecal and intravenous treatments
- e
Adherence to care standards established for SMA
- f
Presence of extraordinary circumstances potentially affecting efficacy or assessment of treatment response.
While the panel agreed that there was a need for measures to control pharmaceutical expenditure, no consensus was reached on which is the most effective measure, with outcome-based payment being the most popular option (56% of votes).
DiscussionThe emergence of new disease-modifying therapies has led to a profound change in the natural history of SMA and a considerable challenge for healthcare systems, both due to the high costs of these treatments and due to the need for constant updates for the specialists responsible for treating and following up these patients. In this context, and given the lack of efficacy data on the new treatments (especially in certain phenotypes), there have been considerable differences between countries in their commercialisation and use. In Spain, the use of nusinersen (the only drug commercialised to date) is regulated by a protocol developed in 2018 by the Ministry of Health with the participation of paediatric neurologists, enabling quick, equitable, and reasonably generalised access to the treatment.9 However, our understanding of the disease and the new treatments has increased considerably since 2018. The lack of a needed update to the protocol led to the creation of a group of Spanish experts in SMA with a view to issuing recommendations to aid in decision-making regarding the use of these new treatments and the evaluation of treatment response.9
SMA is an extremely heterogeneous disease, both genetically (number of SMN2 copies, SMN1 point mutations, polymorphisms with modifying effects in various genes, etc) and clinically (age, functional status, contractures, etc).1 All these characteristics may influence the presentation and course of the disease, as well as the means of evaluating the efficacy of these new treatments; thus, patients must be stratified at least according to age and baseline functional status (presymptomatic, walkers, sitters, non-sitters). In paediatric patients, the maximum motor developmental milestone achieved (SMA types 0-3) and the number of SMN2 copies, the main prognostic factors used before the new treatments were developed, continue to be relevant. However, any stratification may be insufficient for decision-making in specific cases, as there is also a need to take into account some variables that are difficult to predict and/or quantify, such as intercurrent transient processes, social and family setting, quality of life, and life expectancy. In this regard, the implementation of any protocol should be assessed individually for each patient and context, seeking to ensure rational use of the new treatments and an appropriate balance of potential risks and benefits. Therefore, the physician responsible for each patient must have sufficient training and experience in this field. There is also a need for national and/or regional mechanisms (expert committees) to evaluate and resolve cases in which the clinical judgement of the physician responsible for the patient diverges from the recommendations of the relevant treatment protocol.
SMA is characterised by progressive weakness affecting various bodily functions, some of which are vital. Thus, treatment objectives should include a wide range of areas (motor, bulbar, and respiratory function; quality of life) and time periods (short, medium, and long term). The clinical heterogeneity of the disease and in particular the patient’s baseline functional status and the natural history of the disease should also be taken into account. The natural history of the disease is fundamentally influenced by age, disease progression time, and the number of SMN2 copies.10 For example, an initial, more or less prolonged period in which the patient achieves motor developmental milestones may be followed by an initially acute and subsequently slower phase of loss of functional capacity.11 Therefore, while the general objective should be to achieve improvements in one or more clinical areas, short-term stabilisation of some areas may also be a valid objective in patients whose clinical history would lead us to expect functional worsening.2 Furthermore, long-term stabilisation of a progressive, degenerative disease may be considered in general terms to constitute treatment response. However, in addition to efficacy, it is also important to consider the safety of treatments; safety may vary in each patient and as a function of the administration route and the experience of the physician.12 Therefore, we must always aim to achieve a favourable risk-benefit balance in each patient and each therapeutic context.
The clinical heterogeneity of SMA also hinders the evaluation of treatment response; as a result, the types of tools used will vary between patients, mainly in accordance with age and functional status.13 While several specifically designed tools to measure motor function in patients with SMA are now available, these do present certain limitations (the need for specialised staff and materials, prolonged assessment time, patient collaboration, etc) and shortcomings (poor sensitivity, floor and ceiling effects),14,15 particularly in adult patients, limiting their ability to detect changes. Therefore, it is essential also to apply other tools, particularly in the assessment of less well-studied phenotypes, such as in adult patients and those with very poor functional status, to study patient-reported outcomes.2,16,17 Patient-reported outcomes constitute an important complementary tool, as they can be more sensitive than motor scales in detecting small changes, which is particularly relevant in patients with more severe disease.18 They are also able to simultaneously study several aspects, including quality of life, and directly incorporate the patient’s perceptions. Several functional scales are now available that are specifically designed for the assessment of patients with SMA; these include the EK2 scale19,20 and the SMAFRS scale,13 as well as the ALSFRS-R scale,21,22 which was recently validated for use in these patients (data submitted for publication). While they are not applicable to all patient groups (eg, infants), these scales should be adopted in everyday practice due to their clinical relevance, simplicity, and reproducibility. However, respiratory function must be evaluated systematically, particularly in non-walkers and even in the absence of symptoms, in order to ensure the early detection of complications and to assess the need for ventilatory support or assisted coughing.23–25 It is also important for the assessment of bulbar function (speech, salivation, swallowing), particularly among non-sitters.26–30
The criteria regulating the use of nusinersen in the 2018 Spanish Ministry of Health protocol are based on the eligibility criteria of pivotal trials for the drug,31,32 which constituted the best available evidence at the time. However, research in this field has advanced very rapidly and efficacy and safety data have since been reported on the use of nusinersen in other phenotypes, including presymptomatic individuals with 2-3 SMN2 copies33 and adult patients.34,35 There is also a growing body of evidence indicating that baseline functional status is the main factor determining response to the new treatments, with a greater effect than age or number of SMN2 copies.31–35 This has 2 significant consequences for indication of the treatment. Firstly, disease-modifying therapy must be started early, even before symptom onset. Secondly, in the case of symptomatic patients, baseline functional status (rather than the number of SMN2 copies, age, or type of SMA) is the fundamental factor to consider in assessing the potential benefit of a treatment. As a result, in paediatric patients, disease-modifying treatments should be started as early as possible in symptomatic patients, particularly in the first years of life, with the exception of patients presenting symptoms in the first 2 weeks of life (SMA types 0 and 1A), with regard to whom there is no consensus on how to proceed due to the poor vital prognosis despite early treatment.36,37 Furthermore, it is essential to implement neonatal screening for SMA and to start treatment in the presymptomatic stage in children with 1-3 SMN2 copies in order to improve the cost-effectiveness of these high-impact treatments.38 Due to a lack of data, we are currently unable to issue a recommendation on the use of disease-modifying treatments in presymptomatic patients with 4 SMN2 copies.39,40
In the opinion of the panel of experts, continuity of treatment should depend on whether the individually established treatment objectives are achieved, taking into account the expected progression of the disease in each patient. Before determining eligibility or indicating suspension of the treatment, it is essential to rule out acute concomitant factors that may be contributing to transient, potentially reversible worsening. In patients not responding to one treatment during the period established for each patient profile, the drug may be switched for another of the approved treatments, as long as the patient continues to meet criteria for indication of the drug.
Given the heterogeneity of the disease and the continuous research progress being made, deciding which patients should not receive the new treatments or when treatment should be suspended due to ineffectiveness is a complex process, and these decisions should be made on an individualised basis by multidisciplinary teams, always taking into account the best available evidence at the time. New evidence may conflict with excessively rigid interpretations of a protocol that should be considered no more than a guide for clinical decision-making. Decision-making also requires consensus with patients or their parents/guardians. There may be cases in which patients or their parents/guardians disagree with the opinions of the medical team; this demonstrates the vital importance of fluid communication between all parties and of providing the best available information at all times. Patients’ associations, national and regional expert committees, and bioethics committees may help to resolve the most controversial cases, always taking into account the view of the clinician responsible for the patient.
Finally, the expert panel agreed that there was a need to establish measures to control pharmaceutical expenditure for high-impact drugs. However, there was no consensus regarding the most effective measure, although outcome-based payment and a budget ceiling were the most popular options. It should be noted that 3 drugs are currently approved by the European Medicines Agency, each with different dosing; therefore, the most appropriate measure may vary between drugs. Furthermore, there was consensus between participants that physicians should be responsible for ensuring rational use of these treatments, with their indication guided by risk-benefit analysis. Therefore, regarding expenditure, the competent healthcare authorities should determine whether additional measures are needed, after setting prices.
Sufficient consensus was not reached for many issues. Future studies should address these areas of uncertainty as more evidence comes to light. These issues include recommendations on which drugs are most appropriate for different patient profiles, and how to switch between or combine different treatments.
In conclusion, these recommendations are intended to be used as a framework to assist clinicians and regulators in the appropriate use of disease-modifying treatments for SMA.
Conflicts of interestI. Pitarch Castellano has received fees for consulting and training activities from Biogen, Roche, and Novartis.
M. Cabrera-Serrano has received fees from Biogen for scientific translations.
R. Calvo Medina has received fees for participating in courses and training activities from Biogen, Roche, and Novartis.
S. Espinosa García has received fees for participating in courses and training activities from Biogen, Roche, and Novartis.
J.A. Fernández-Ramos has received fees for participating in training activities from Biogen and Novartis.
O. García Campos has received fees for consulting and training activities from Biogen, Roche, Genzyme, and PTC.
D. Gómez-Andrés has received fees for consulting and training activities from Biogen.
M.A. Grimalt Calatayud has received fees for participating in courses and training activities from Biogen.
A.J. Gutiérrez Martínez has received fees for participating in courses and training activities from Biogen.
E. Ibáñez Albert has received fees for consulting and participating in courses from Biogen.
S. Kapetanovic García has received fees for consulting and training activities from Biogen.
M. Madruga-Garrido has received fees for participating in research projects (including clinical trials), committees, courses, and training activities from Biogen, Roche, and Novartis.
M. Martínez-Moreno has received fees for participating in committees, courses, and training activities from Biogen, Roche, and Novartis.
J. Medina Cantillo has received fees for participating in research projects (including clinical trials), courses, and training activities from Biogen, Roche, and Novartis.
A. Moreno Escribano has received fees for participating in training activities from Biogen.
F. Munell has received fees for consulting and training activities from Biogen, Roche, and Novartis.
A. Nascimento Osorio has received fees for participating in research projects (including clinical trials), committees, courses, and training activities from Biogen, Roche, and Novartis.
S.I. Pascual-Pascual has received fees for participating in clinical trials, committees, courses, and training activities from Biogen, Roche, and Novartis.
M. Povedano has received fees for consulting, courses, and training activities for Biogen and Roche.
J.F. Vázquez-Costa has received fees for consulting, courses, and training activities from Biogen and Roche.
The remaining authors have no conflicts of interest to declare.
Please cite this article as: Pitarch Castellano I, Cabrera-Serrano M, Calvo Medina R, Cattinari MG, Espinosa García S, Fernández-Ramos JA, et al. Consenso Delphi de las recomendaciones para el tratamiento de los pacientes con atrofia muscular espinal en España (consenso RET-AME). Neurología. 2022;37:216–228.
