




La enfermedad de Creutzfeldt-Jakob (ECJ) es una encefalopatía priónica con combinación variable de demencia, ataxia, mioclonías y signos piramidades y extrapiramidales. Las enfermedades priónicas pueden presentarse de forma esporádica, familiar o adquirida. Las encefalopatías espongiformes transmisibles (EET) esporádicas son el 85% de los casos e incluyen la enfermedad de Creutzfeldt-Jakob esporádica (ECJe) y el insomnio fatal esporádico. Las EET familiares o genéticamente determinadas son el 10-15% de los casos y comprenden la enfermedad de Creutzfeldt-Jakob familiar (ECJf), el insomnio fatal familiar y la enfermedad de Gerstmann-Scheinker. Las EET adquiridas (< 5% de los casos) incluyen el kuru, la enfermedad de Creutzfeldt-Jakob iatrogénica y la enfermedad de Creutzfeldt-Jakob variante (ECJv)1. El patrón oro diagnóstico de las enfermedades priónicas es el estudio anatomopatológico. Los hallazgos histopatológicos clásicos de la ECJ son la pérdida de neuronas, gliosis y vacuolización (antes llamado cambios espongiformes), siendo necesario detectar mediante inmunotinción la proteína PrPSc (proteína priónica scrapie)2. Los criterios vigentes utilizados para el diagnóstico de enfermedad de Creutzfeldt-Jakob datan del año 2003 por la OMS3.
Existen descritos en la literatura médica casos clínicos que simulaban una ECJ como son la encefalopatía de Wernicke, leucoencefalopatía multifocal progresiva, encefalopatía asociada a anticuerpos CKGV, angiopatía amiloide, encefalopatía de Hashimoto, el síndrome de Gerstmann-Sträussler-Scheinker, encefalopatía inducida por litio, ataxia cerebelar por anticuerpos anti-Gad, gliomatosis cerebral, meningitis carcinomatosa y Alzheimer rápidamente progresivo entre otros (tóxicos, carenciales, metabólicos, vasculares, infecciosos, autoinmunes, neoplásicos, psiquiátricos, neurodegenerativos, ect.)1. Presentamos un caso de demencia rápidamente progresiva con síntomas piramidales y extrapiramidales, mutismo y ataxia por déficit de cianocobalamina (B12) que simuló una probable ECJe.
Se trata de una mujer de 64 años con antecedentes de zóster intercostal y posterior cuadro de astenia con pérdida de peso de 2 años de evolución que no fueron filiados pese a diversos estudios. Consultó por cuadro de deterioro cognitivo progresivo de 2 meses de evolución con inatención, desorientación, lenguaje poco fluido y solo ejecutaba órdenes sencillas. Presentó una distonía cervical (laterocolli izquierdo) y de miembros superiores, una paraparesia espástica con piramidalismo y ataxia en extremidades. Durante los primeros días de ingreso progresó a encamamiento y mutismo.
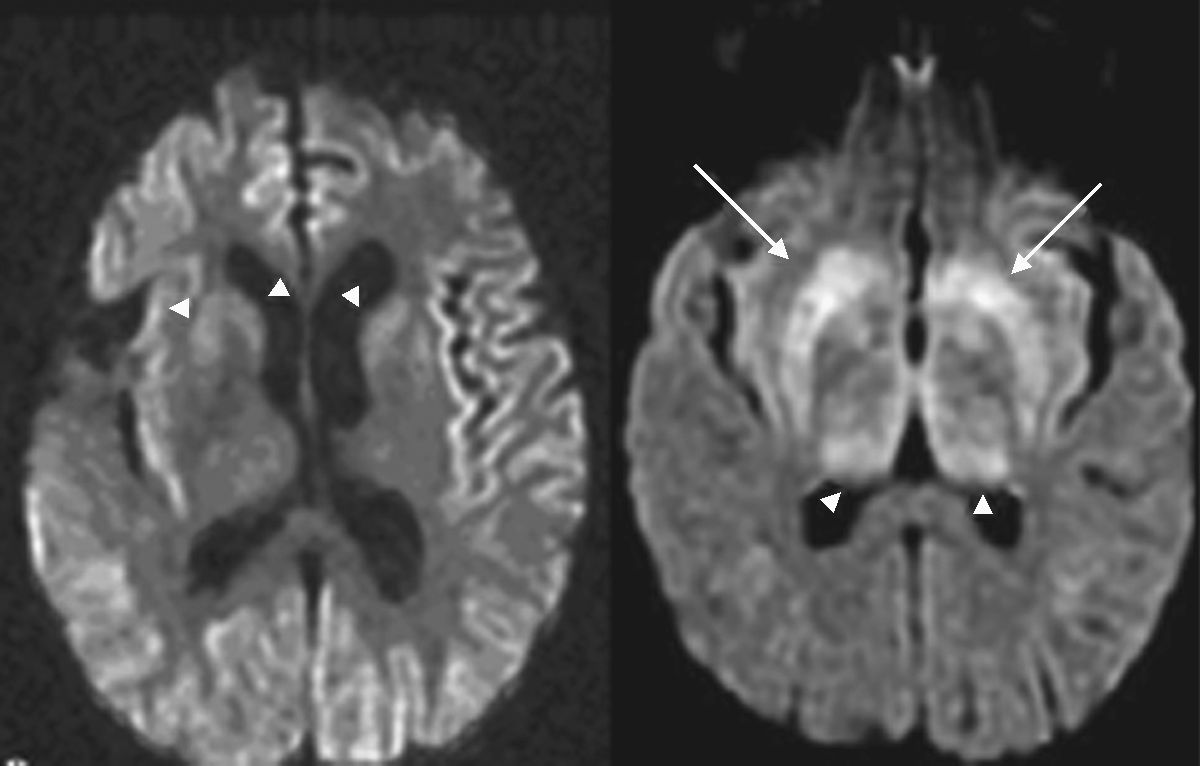
El electroencefalograma mostró ondas periódicas lentas y la RMN craneal una hiperintensidad en ganglios basales, talámica medial bilateral y pulvinar en secuencia de Difusión, además hiperintensidad en vía piramidal y extrapiramidal en T1, FLAIR y Difusión (figs. 1 y 2). Las serologías de hepatitis y VIH, Ac anti-GAD, anti-Hu/Ma2, antineuronales, antitiroideos, hormonas tiroideas y proteína 14-3-3 fueron negativos o normales. Se encontró déficit de vitamina B12 (20 pg/ml, N<223), anticuerpos antifactor intrínseco positivos y gastrina elevada. Se realizó biopsia gástrica con resultado de gastritis crónica en cuerpo y antro.

, FLAIR y Difusión (fila inferior derecha).")
Se inició tratamiento con cianocobalamina con una remisión progresiva parcial, más significativa en el plano motor aunque también en el plano cognitivo. Desapareció por completo la distonía cervical y de hombros con disminución del dolor que le producían. Comenzó la deambulación precisando ayuda bilateral con una marcha atáxica con lateralización hacia la derecha, además leve dismetría en la maniobra dedo-nariz. Desde el punto de vista cognitivo persistió el déficit de atención, desorientación temporoespacial y fallos de memoria inmediata. Mejoró su nominación (6/8 animales), su comprensión de órdenes sencillas y la repetición de frases. Tras 3 meses de tratamiento recuperó su situación basal sin secuelas tanto en el plano motor como cognitivo, pudiendo mantener una conversación normal.
Existen varios criterios clínicos diagnósticos de ECJ como son los criterios vigentes de la OMS 2003 y los criterios Masters3,4. Los criterios de la OMS probablemente sean los más utilizados3 (tabla 1). Las cifras de sensibilidad y especificidad de la prueba de detección de proteína 14-3-3 para el diagnóstico de EJCe oscilan entre 88-97% y 84-100%, respectivamente4. La detección de 14-3-3 puede resultar negativa en las fases iniciales de la enfermedad y hacerse positiva en las fases posteriores teniendo su mayor utilidad cuando los resultados concuerdan con la sospecha clínica, consiguiendo en estos casos incrementar las razones de verosimilitud en comparación con los datos clínicos aislados5,6.
Criterios OMS 2003 (enfermedad de Creutzfeldt-Jakob esporádica).
| ECJe posible |
| Demencia progresiva más duración menor de 2 años más los estudios no sugieren otro diagnóstico más 2 o más de los 4 siguientes: |
| 1. Mioclonías |
| 2. Signos o síntomas visuales o cerebelosos |
| 3. Signos o síntomas piramidales o extrapiramidales |
| 4. Mutismo acinético |
| ECJe probable |
| ECJe posible más al menos uno de los siguientes criterios: |
| 1. Complejos periódicos EEG con cualquier duración de la enfermedad |
| 2. Proteína 14-3-3 en casos con supervivencia menor a 2 años |
| ECJ definitiva |
| Confirmación patológica y/o presencia de PrP resistente a proteasas (inmunohistoquímica o Western blot) y/o presencia de fibras asociadas a scrapie (ME) |
ECJe: enfermedad de Creutzfeldt-Jakob esporádica; ME: microscopia electrónica; PrP: proteína priónica.
Los criterios Masters requieren una demencia progresiva con 2 de los siguientes: mioclonías, piramidal, extrapiramidal, cerebelosa y EEG típico con descargas epileptiformes periódicas focales aproximadamente 1Hz o difusas7. El patrón característico de la ECJe consiste en complejos periódicos que aparecen sobre una actividad de fondo enlentecida de forma difusa3. Los valores de sensibilidad y especificidad para el diagnóstico de ECJe oscilan entre 58-67% y el 74-91% respectivamente5. Aunque los complejos suelen distribuirse de forma generalizada, en las fases iniciales pueden limitarse a un hemisferio o incluso a una región8. El diagnóstico diferencial de un patrón EEG tipo ECJ incluye una variedad de procesos degenerativos (enfermedad de Alzheimer, demencia con cuerpos de Lewy), vasculares (enfermedad de Binswanger, anoxia), infecciosos (abscesos múltiples, demencia del sida, panencefalitis esclerosante subaguda), tóxicos (ketamina, fenciclidina, baclofeno, mianserina, metrizamida, litio) y metabólicos (encefalopatía anóxica, hiperamonemia, MELAS). Este patrón debe diferenciarse asimismo de otras actividades periódicas como las descargas epileptiformes lateralizadas periódicas y el estatus epiléptico1,9.
Además, la resonancia magnética cerebral representa uno de los mayores avances para el diagnóstico de la EET obtenidos en los últimos años. Los hallazgos característicos de la ECJ consisten en una hiperintensidad de señal localizada en el estriado, la corteza cerebral o ambos. Las lesiones pueden ser unilaterales o bilaterales y parecen instaurarse de forma precoz, incluso a las 3 semanas del inicio de los síntomas. Los valores de sensibilidad y especificidad para el diagnóstico de ECJe utilizando secuencias FLAIR y DWI oscilan entre el 80-100% y el 94-100% respectivamente1. El déficit de cianocobalamina se puede traducir en cambios cognitivos, trastornos afectivos, degeneración combinada subaguda, neuropatía periférica y atrofia óptica entre otros10.
En definitiva, nuestra paciente cumplía criterios OMS 2003 de ECJe probable con una demencia progresiva menor de 2 años, 3 de 4 criterios clínicos (signos cerebelosos, signos-síntomas piramidales/extrapiramidales y mutismo acinético) y además complejos periódicos en EEG. La determinación de 14-3-3 fue negativa, el déficit severo de B12 y la mejoría clínica tras tratamiento cambiaron el diagnóstico. El déficit de cianocobalamina secundario a gastritis atrófica por su comportamiento clínico y hallazgos electroencefalográficos podría en ocasiones simular una ECJ.

