



El vasoespasmo (VSP) ha sido tradicionalmente considerado como el principal determinante de mal pronóstico tras sufrir una hemorragia subaracnoidea (HSA). Como consecuencia, la mayoría de las líneas de investigación y los tratamientos están dirigidos hacia la reducción de la incidencia de dicho VSP. Hasta la fecha, sin embargo, los resultados de los ensayos clínicos basados en esta estrategia no se han traducido en un tratamiento definitivo capaz de prevenir o mejorar la lesión cerebral tras una HSA. Este hecho ha provocado un cambio de paradigma en el interés investigativo, focalizándolo hacia la lesión cerebral precoz (LCP), que se produce en las primeras 72 h tras la HSA. Así mismo, ha modificado la visión que se tenía de la responsabilidad del VSP sobre el daño cerebral y sugiere la necesidad de una re-evaluación del proceso fisiopatológico de la HSA.
DesarrolloEsta revisión examina el estado actual del conocimiento de los mecanismos fisiopatológicos relacionados con la LCP y resume las opciones diagnósticas disponibles en la actualidad.
ConclusiónParece necesario cambiar la dirección en la investigación de esta enfermedad, centrándose en la prevención de la LCP, la reducción de las complicaciones cerebrales secundarias y en última instancia, la optimización de los resultados neurológicos.
Delayed vasospasm has traditionally been considered the most important determinant of poor outcome after subarachnoid haemorrhage (SAH). Consequently, most of the research and therapies are directed towards reducing the incidence of vasospasm (VSP). To date, however, clinical trials based on this strategy have not delivered a definitive treatment for preventing or reducing brain injury after SAH. This fact has caused a paradigm shift in research, which now focuses on early brain injury (EBI) occurring in the first 72hours after SAH. It has also changed the idea of VSP's role in brain damage, and suggests the need for re-evaluating the pathophysiological process of SAH.
DevelopmentThis review examines the current state of knowledge on the pathophysiological mechanisms associated with EBI and summarises the diagnostic options currently available.
ConclusionIt seems that the research approach needs to be changed so that investigators will focus on prevention of EBI, reduction of secondary brain complications and ultimately, the optimisation neurological outcome.
La hemorragia subaracnoidea (HSA) continúa siendo una de las enfermedades neurológicas de mayor interés, siendo causa del 5% del total de los casos de infarto cerebral1. Alrededor de un 10-12% de los pacientes fallecen antes de llegar al hospital y un 45-50% lo hacen a lo largo de los 30 primeros días2. De los pacientes que sobreviven, el 30% presenta secuelas invalidantes y el 66% afirma no mantener la misma calidad de vida que tenían antes del proceso2.
En el pasado, y basándose en la correlación clínica existente entre la aparición del vasoespasmo (VSP) y el deterioro neurológico, la investigación se ha centrado en limitar o impedir la vasoconstricción arterial en un intento de combatir la alta morbimortalidad asociada a la HSA. Mientras nuestros conocimientos acerca de la fisiopatología del VSP han progresado significativamente, esta comprensión no se ha traducido en la obtención de tratamientos clínicamente eficaces. Resultados como los obtenidos por el clazosentan3 han modificado la visión de la responsabilidad del VSP sobre el daño cerebral tras una HSA, sugiriendo así, la necesidad de una revaluación sobre su proceso fisiopatológico y afirmando que tanto el VSP como sus consecuencias clínicas no deben ser reconocidas como las únicas causas de mal pronóstico tras una HSA. Es más, la literatura ratifica que la presencia de VSP no es un prerrequisito para la aparición de daño cerebral tardío o de mal pronóstico clínico tras una HSA4,5.
En los últimos años, se han analizado dos conceptos fundamentales desarrollados a lo largo de la fase previa al VSP: la lesión cerebral precoz (LCP) y la despolarización cortical propagada (DCP)6,7. Tanto a nivel experimental como clínico, se ha señalado la importancia de este período, reconociendo el significativo papel jugado por la isquemia transitoria que sucede al inicio de la HSA, la alteración de la barrera hematoencefálica (BHE) y la detección de isquemia cortical generalizada tras el sangrado. Existen evidencias de que los eventos fisiológicos y celulares de la LCP, que se producen durante las primeras 72h tras la rotura del aneurisma, contribuyen significativamente en la evolución de los pacientes, pudiendo ser incluso considerados factores más importantes en el pronóstico de esta enfermedad que el propio VSP8. Por tanto, la LCP debe ser considerada como objetivo principal en la investigación de la HSA.
La presente revisión analiza el estado actual del conocimiento acerca de los mecanismos fisiopatológicos asociados a la LCP y resume las opciones diagnósticas disponibles en la actualidad.
DesarrolloLa lesión cerebral precoz y sus mecanismos fisiopatológicosUno de los avances más importantes acaecidos en los últimos años es el reconocimiento de la aparición de la LCP tras la HSA, las consecuencias derivadas de la hemorragia inicial y su efecto perjudicial sobre el resultado clínico del paciente. El término LCP ha sido recientemente acuñado y se refiere a la lesión cerebral que se produce dentro de las primeras 72h tras la HSA, antes del desarrollo del VSP.
Estudios recientes han focalizado su atención sobre el uso de agentes terapéuticos capaces de mejorar la LCP. Ejemplos de ello son las estatinas9 —capaces de atenuar la vía pro-apoptótica dependiente—, la melatonina10 —neurohormona con capacidad antioxidante que mitiga el edema cerebral e incrementa la supervivencia en modelos experimentales— o el óxido hiperbárico11 —que mejora la LCP post-HSA experimental al contrarrestar los efectos nocivos de la DCP y regular los genes de sistemas proteicos implicados en la respuesta frente al estrés oxidativo. Datos que apuntan de forma indirecta a la participación de diferentes vías y mecanismos involucrados en la fisiopatología de la LCP y que obliga a conocer e indagar más profundamente la fisiopatología de la LCP.
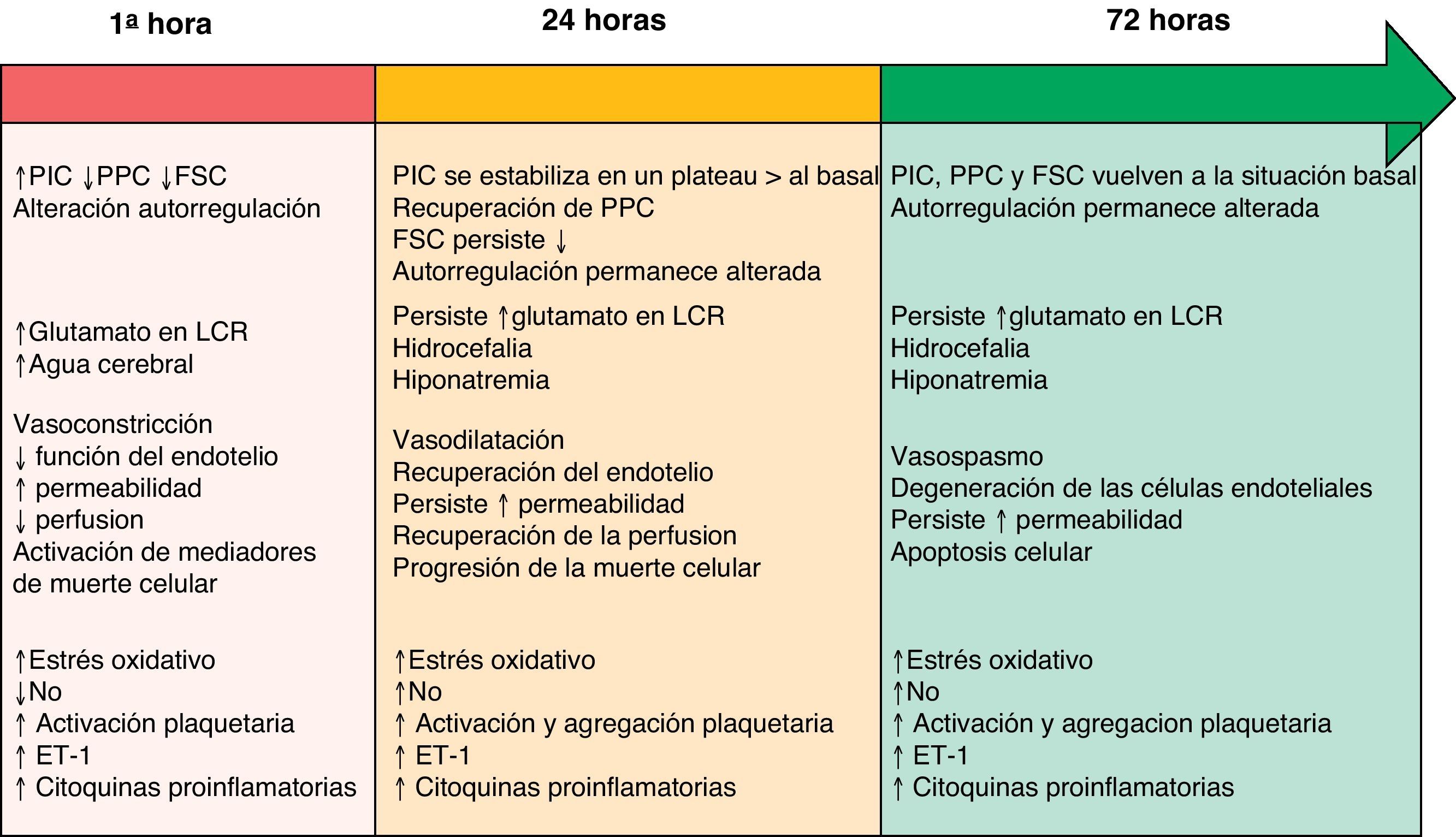
Aunque separados en este manuscrito por apartados con el fin una mejor comprensión, existe una clara interrelación y simultaneidad entre los distintos mecanismos fisiopatológicos descritos a continuación. Su línea evolutiva temporal durante la fase de LCP está resumida en la figura 1.

Línea evolutiva temporal de los diversos mecanismos fisiopatológicos durante la fase de LCP tras una HSA. Modificado de Sehba et al.26
ET-1: endotelina-1; FSC: flujo sanguíneo cerebral; LCR: líquido cefalorraquídeo; NO: óxido nítrico; PIC: presión intracraneal; PPC: presión de perfusión cerebral.
El daño cerebral inicial se produce como consecuencia del efecto mecánico causado por la salida de la sangre al espacio subaracnoideo3,12. Este traumatismo provoca la constricción y compresión de las arterias adyacentes al foco de sangrado, así como el desplazamiento del líquido cefalorraquídeo (LCR). Como consecuencia de ello se produce un aumento generalizado de la presión intracraneal (PIC), cuya intensidad está relacionada con el volumen de sangre, la obstrucción a la salida del LCR y la difusión de la vasodilatación arterial cerebral distal13. La severidad de la PIC está asociada a cambios en el metabolismo cerebral, inflamación, caída del flujo sanguíneo cerebral (FSC, atribuido al breve periodo non-reflow generado por la elevación de la PIC y el descenso de la presión de perfusión cerebral [PPC]) y el desarrollo de isquemia cerebral4,14,15. De manera que sus niveles son, a menudo, utilizados como predictores del pronóstico de la HSA16. A medida que pasan las primeras horas tras la hemorragia, la PIC y la PPC sufren fluctuaciones hasta alcanzar horas/días después sus niveles basales17. Sin embargo, el FSC puede recuperarse o permanecer disminuido, y la capacidad de autorregulación cerebral persiste alterada a las 72h del inicio de la clínica18.
Aunque los vasos cerebrales de gran tamaño contribuyen a la resistencia vascular, no afectan significativamente al FSC19. Si las grandes arterias se contraen y las arteriolas se dilatan, se produce un descenso de la presión microvascular sin cambios en el FSC, mostrando cómo dicha presión está regulada de manera independiente y contradiciendo el concepto que se tiene de que los grandes vasos contribuyen de forma importante en el descenso del FSC. Recientemente, han sido identificados cambios en la estructura anatómica de los microvasos cerebrales suficientes como para provocar deficiencias funcionales tras una HSA20. Se piensa que la función de autorregulación vascular cerebral falla para compensar las reducciones del diámetro vascular a nivel de los microvasos, aumentando de esta forma el riesgo de isquemia. Estos cambios pueden explicar la génesis de isquemia cerebral en humanos en ausencia de evidencia angiográfica de vasoconstricción en vasos de gran tamaño21.
InflamaciónLa inflamación es una reacción causa-efecto propia de los tejidos vivos ante cualquier lesión. La sangre extravasada por una HSA sería la responsable de la activación de una cascada de reacciones que llevan a la producción de varios factores vasoactivos y proinflamatorios en el espacio subaracnoideo. La activación de la cascada inflamatoria tras la HSA incluye moléculas de adhesión (VCAM-1, ICAM1 y E-selectina) necesarias para la acumulación de leucocitos a nivel de los tejidos inflamatorios, citocinas (IL-6, IL-1, TNF-α) y factores del complemento (C3a y C5a), capaces de acelerar la lisis eritrocitaria y, en consecuencia, liberar los factores espasmogénicos de dichas células22. Estudios clínicos en los que se han utilizado agentes antiinflamatorios23 han demostrado el carácter multifactorial de la inflamación post-HSA. Actualmente, se realizan estudios en los que se manejan antagonistas específicos de IL y anticuerpos con el fin de bloquear las interacciones leucocito-endoteliales.
MicrotrombosisLa activación plaquetaria se produce a los pocos minutos de la HSA, promoviendo un mecanismo inflamatorio adicional, capaz de agravar la lesión cerebral secundaria. La mayoría de los perfiles de perfusión vascular reducida observada en estudios experimentales24,25, presentaron agregados plaquetarios y pérdidas focales del colágeno IV. Estos datos demostraron que los microvasos del parénquima tienen comprometida su función a los 10min tras el inicio de la HSA e identificaron la constricción microvascular focal y la acumulación local de agregados plaquetarios luminares como potenciales estímulos de tal compromiso.
Estrés oxidativoEl estrés oxidativo se define como la situación que ocurre cuando el equilibrio fisiológico entre oxidantes y antioxidantes se ve alterado en favor de los primeros, dando lugar a un daño molecular, y por consiguiente celular, por oxidación. Estudios recientes8,26 han proporcionado pruebas suficientes de que el estrés oxidativo desempeña un papel importante en la LCP. Las fuentes más importantes de generación de radicales libres (ROS) tras una HSA son: la liberación del anión superóxido, estimulada por la presencia de oxihemoglobina en el LCR, y el peróxido de hidrógeno, producido tras la oxidación de la hemoglobina a metahemoglobina27.
Los ROS provocan daño en el endotelio, en la musculatura vascular lisa, alteran la BHE e inducen apoptosis celular tras una HSA4,28. Por otro lado, la deficiencia de efectividad de los sistemas antioxidantes endógenos durante la HSA parece contribuir al deterioro evolutivo neurológico de estos pacientes. Prueba de ello son los trabajos realizados en los que el uso de terapia antioxidante ha proporcionado neuroprotección tras una HSA, preservando la BHE29, evitando la muerte celular30 y mejorando las puntuaciones neurológicas finales26.
Óxido nítricoEl óxido nítrico (NO) desempeña un papel fundamental dentro de la hemodinámica cerebral, de ahí que cualquier alteración en sus niveles conlleve consecuencias patológicas variables. El NO es producido por las tres isoformas de óxido nítrico sintetasa (NOS; neuronal, endotelial e inducible) y tiene una vida media segundos. Los niveles de NO descienden, se recuperan y aumentan a lo largo de las primeras 72 h tras una HSA31. Este rango tan variable de niveles da lugar a cambios que van desde la vasoconstricción, descenso del FSC, agregación plaquetaria y adhesión leucocitaria durante las primeras horas, hasta la aparición de daño neurológico isquémico tardío y peor pronóstico clínico32.
EndotelinaLa endotelina-1 (ET-1), factor de crecimiento derivado de las plaquetas y citocinas proinflamatorias, es un potente vasoconstrictor liberado a partir de los leucocitos y astrocitos en respuesta a la presencia de inflamación e isquemia precoz tras una HSA33. Muchos estudios han demostrado niveles elevados de ET-1 en plasma, LCR y microdializado tras una HSA34. Su liberación se produce a los pocos minutos del inicio del sangrado, aumentando rápidamente sus niveles al tiempo que se reducen los de NO, provocando cambios morfológicos degenerativos a nivel de la pared vascular. Esta situación favorece la vasoconstricción cerebral y provee otro mecanismo relacionado con la patogénesis del vasoespasmo tardío y el daño neurológico isquémico tardío35.
Despolarización cortical propagadaLa HSA provoca una modificación en la homeostasis iónica, favoreciendo la vasoconstricción y aparición de alteraciones en la actividad eléctrica, como la denominada DCP. La DCP describe una onda de despolarización de la masa neuronal asociada con la afluencia neta de cationes (sodio y calcio) y agua. Si la homeostasis iónica no es reestablecida, el edema celular persiste en el tiempo (edema citotóxico) pudiendo inducir la muerte celular. La DCP provoca modificaciones en el tono vascular dando lugar a áreas de hiperperfusión transitoria en el tejido sano (respuesta hemodinámica fisiológica) mediante dilatación vascular, o bien, a áreas de hipoperfusión en el tejido en riesgo de lesión progresiva (respuesta hemodinámica inversa) al provocar un espasmo severo a nivel de los microvasos36. En este segundo caso, el déficit de perfusión propagado prolonga la situación de despolarización neuronal. Estudios experimentales han evidenciado que tanto la oxihemoglobina como la elevación extracelular de potasio, el descenso de NO, glutamato y ET-1, están implicados en el desarrollo de la DCP tras una HSA37,38. Estudios clínicos recientes39,40 proporcionan directa e inequívocamente evidencias electrofisiológicas acerca de la existencia de la DCP en la HSA y su relación con el desarrollo de la lesión isquémica tardía.
Hiponatremia e hipomagnesemiaLa incidencia publicada de la hiponatremia tras HSA se encuentra alrededor del 10-30%1. Su aparición es más frecuente en pacientes con mala situación clínica inicial, aneurismas de la arteria comunicante anterior e hidrocefalia y está considerada como factor de riesgo independiente de mal pronóstico.
Por lo que respecta al magnesio, aproximadamente un 38% de los pacientes con HSA presentan niveles disminuidos a las 48 h tras el inicio de la patología. Está relacionado con la dilatación vascular y la inhibición de la agregación plaquetaria y de la síntesis de ET-1, de forma que su reducción exacerba la LCP. Así mismo, estos niveles reducidos contribuyen al incremento del calcio intracelular, considerado como el mediador predominante de la muerte neuronal. Su conocimiento está abriendo nuevos horizontes terapéuticos, como el estudio MASH-II41, en actual proceso.
GlutamatoSe ha observado que el glutamato y los transportadores de glutamato (GT) tienen un papel importante en la patogénesis de la lesión neurológica isquémica. Sus niveles parecen estar relacionados con la intensidad del daño inicial. Los cambios en el glutamato, GT y el daño neuronal tras una HSA aún no han sido ampliamente investigados. Sin embargo, la HSA inducida en un modelo de rata parece producir un aumento excesivo y prolongado de las concentraciones de GT extracelular y regulación a la baja de GT, que se acompañan de un aumento del espesor de pared de las arterias basilares y la degeneración neuronal del hipocampo42.
Apoptosis celularEl fenómeno inicial acaecido inmediatamente después de la rotura del aneurisma es la detención aguda del FSC, circunstancia que da lugar a una situación de isquemia cerebral global transitoria que puede ser letal por sí sola. En aquellos casos en los que el paciente sobrevive a este evento, puede aparecer un insulto isquémico secundario debido a la alteración de la BHE, capaz de progresar hacia la formación de edema cerebral generalizado y/o apoptosis celular. El edema cerebral contribuye a un nuevo aumento de la PIC y, por tanto, a una mayor reducción del FSC43. El mecanismo de lesión de la BHE no está claro, pero la apoptosis celular se ha postulado como una de sus posibles causas44. La alteración de dicha BHE y la consiguiente formación de edema cerebral han sido señaladas como los principales factores predictivos de disfunción cognitiva tras una HSA45. La apoptosis celular ha sido implicada incluso en la formación y rotura del aneurisma, tanto en modelos animales como en humanos46,47. Entre las diversas vías de activación de la apoptosis celular estudiadas, tres han sido reconocidas como principales6: la vía del receptor de mortalidad (Fas, TNFR1 y DR 3-5), la vía caspasa-dependiente (representada por las caspasas 3 y 8) e independiente (representada por el factor inductor de apoptosis), y la vía mitocondrial (representada por el citocromo c y el factor de transcripción nuclear p-53). Los núcleos diana de estas vías de muerte celular son fundamentalmente las neuronas, las células gliales y la vasculatura cerebral (músculo liso y endotelio).
Opciones diagnósticasEs evidente que muchas de las técnicas diagnósticas de HSA actuales son solo capaces de identificar los cambios fisiológicos una vez el daño neuronal está probablemente establecido de manera irreversible. Por tanto, una fracción considerable de la investigación actual está siendo dirigida hacia la identificación de marcadores que puedan permitir una detección temprana de las lesiones derivadas de la HSA. A este efecto, dos son las principales líneas de investigación: técnicas de neuroimagen y marcadores biológicos en fluidos corporales.
Técnicas de neuroimagenA lo largo de los últimos años, han sido utilizados varios métodos de estudio para la medición de la perfusión cerebral48-50 incluyendo, la tomografía computarizada por emisión de positrones, la tomografía computarizada por emisión simple de fotones, la tomografía computarizada con xenón y el Doppler transcraneal (DTC). De todas estas modalidades, el DTC ha sido la más utilizada por sus buenos resultados y ausencia de invasividad. No obstante, es preciso reconocer sus limitaciones: operador dependiente, su incapacidad para cuantificar el FSC y su baja especificidad como guía de la terapéutica a seguir. Por otro lado, las imágenes obtenidas a través de tomografía computarizada están registrándose como «tema candente» en este área y, dentro de sus opciones, destaca la TCP51.
La TCP está siendo ampliamente investigada como medio diagnóstico precoz tanto a nivel de la LCP52 como del VSP cerebral53, con resultados esperanzadores. La TCP puede proporcionar varios parámetros cuantitativos de la hemodinámica cerebral incluyendo, el tiempo de tránsito medio (TTM), el volumen sanguíneo cerebral y el FSC. En la práctica, la inspección visual de los mapas de la TCP puede ser utilizada de manera fiable para evaluar la hipoperfusión cerebral. Dichos mapas pueden mostrar una disminución de la perfusión en zonas con un mínimo o ausencia de VSP objetivable por angiografía, lo que permite el reconocimiento del riesgo isquémico, que de otra manera pasarían desapercibidos. De hecho, los hallazgos obtenidos en diferentes estudios con TCP parecen tener un mayor valor predictivo que los obtenidos mediante TC angiografía51 o DTC54, para el diagnóstico de lesión isquémica cerebral tardía en pacientes con signos de deterioro clínico.
Dentro de las mediciones analizadas por la TCP, el TTM ha sido identificado no solo como el parámetro de perfusión más sensible a la hora de detectar VSP cerebral, sino también como predictor independiente de mortalidad precoz tras una HSA55. En cualquier caso, se adivina como un parámetro prometedor dentro de la fase de LCP tras la HSA, para la detección temprana de aquellos pacientes con alto riesgo de sufrir una mala evolución clínica.
Marcadores biológicosExiste una extensa literatura centrada en el análisis de biomarcadores dentro del contexto de distintas muestras biológicas humanas y la HSA. Atendiendo al momento histórico, han sido empleadas diversas técnicas y perspectivas teóricas. Cada enfoque, ha sido dado sobre la base de la comprensión teórica de datos como los mecanismos fisiopatológicos subyacentes en el desarrollo del VSP o la actual LCP.
Técnicas como la microdiálisis cerebral (MDC) y la proteómica (PT) podrían ser útiles en el estudio de la patogenia de la LCP y el VSP tras una HSA.
La MDC, aunque con resultados contradictorios, se considera una herramienta prometedora para el seguimiento de los pacientes que sufren una HSA. Se ha constatado que las reducciones transitorias del FSC se correlacionan con la elevación extracelular del glutamato y el glicerol, mientras la razón lactato/piruvato es solo sensible tras una hipoperfusión prolongada56. Los ROS, los metabolitos derivados del NO, algunas citocinas y proteínas astrocíticas, han sido rescatados mediante microdiálisis y propuestos como candidatos a biomarcador de VSP57. El futuro éxito de la MDC dependerá en gran medida de la elección de los biomarcadores, su sensibilidad, especificidad y valor predictivo de eventos neuroquímicos secundarios, así como de la disponibilidad de métodos prácticos para el análisis químico de los marcadores individuales.
La PT permite una proyección simultánea y a gran escala de todas las proteínas en una muestra biológica. Esta técnica de detección avanzada también puede acceder a la recolección objetiva, no sesgada, de datos que permitan la identificación de biomarcadores y/o nuevas dianas terapéuticas que pueden no ser intuitivamente vinculadas con alguno de los procesos fisiopatológicos de la enfermedad.
Autores como Lad et al.58, han revisado la evolución de los marcadores biológicos del VSP en el LCR. Según su criterio podrían dividirse en tres categorías atendiendo a su valor predictivo: a) marcadores de gran valor; b) marcadores candidatos, y c) marcadores no candidatos. Dentro de la categoría A encontraríamos: el TNF, el receptor soluble de necrosis tumoral I, el receptor antagonista de la IL-1 y las proteínas neurofilamentosas NFL y NfH. Dentro de la categoría B: la apolipoproteína E, la F2-isoprostano, la NADPH oxidasa y el complejo trombina-antitrombina III como marcadores del pronóstico neurológico, mientras que la E-selectina, el lactato y los productos de degradación de la alfa-II espectrina lo serían para el pronóstico del VSP. La categoría C contendría: la proteína glial S100B, el factor de crecimiento plaquetario, la ICAM-1, la VCAM-1 y la IL-8.
Neuromonitorización multimodalSon cada vez más los autores que afirman que la combinación de datos procedentes de distintas técnicas puede proveer al clínico de mayor información, al ofrecer cada uno de ellos una perspectiva diferente de la fisiología y del metabolismo cerebral. Este concepto, conocido como «neuromonitorización multimodal» ha emergido como un método prometedor dentro del territorio específico de los pacientes con HSA. Ejemplos de ello serían: el uso conjunto de los sistemas de seguimiento convencionales (PIC, DTC, etc.) con la evaluación directa de marcadores bioquímicos de MCD o la combinación de técnicas avanzadas de seguimiento cerebral y cardiovascular59.
ConclusionesLa LCP abre una nueva frontera en la investigación de la HSA, pudiendo ser un colaborador clave de la alta morbimortalidad de esta patología. Esta nueva puerta abierta ha de promover esfuerzos colaborativos entre las neurociencias básicas, la neurocirugía, la neurología y el neurointensivismo con el fin de permitir una prevención más efectiva de las lesiones isquémicas secundarias derivadas de la LCP.
Es probable que la LCP sea el resultado de una serie de mecanismos fisiopatológicos relacionados entre sí, con un mismo resultado: la muerte celular. Está pendiente saber cuál o cuáles de estos factores son clave. Lo único claro es que la investigación está avanzando de forma exponencial y que, teniendo en cuenta el hecho de que la reversión del VSP no parece mejorar los resultados clínicos, puede ser que en el futuro, los tratamientos anti-LCP puedan proporcionar una opción terapéutica viable en los pacientes con HSA.
Conflictos de interesesLos autores declaran que no hay ningún conflicto de interés.
Al Dr. Rafael León López y a Noa León Muñoz, por su inestimable apoyo.

