










Se desconoce la incidencia de la distrofia miotónica tipo1 (DM1), enfermedad con gran variedad fenotípica, en nuestra región. El objetivo de nuestro trabajo es estimar la incidencia de DM1 en nuestro centro (referencia en Aragón) e identificar las características propias de nuestra población (correlación genotipo-fenotipo).
MétodosEstudio descriptivo retrospectivo de 459 pacientes clasificados según número de repeticiones CTG en: normal (5-35), premutado (36-50), protomutado (51-80), pequeñas expansiones (81-150), intermedias (151-1.000) y grandes (>1.000). Además, según el fenotipo mostrado, se categorizaron como: no afectos (5-50CTG), forma leve o asintomática (51-150CTG), clásica (151-1.000CTG) y severa (>1.000CTG).
ResultadosLa incidencia de DM1 fue de 20,61 (IC95%: 19,59-21,63) casos por millón de individuos-año. Se evidenció una correlación inversa entre el número de CTG y la edad al diagnóstico genético (ρ=−0,547; IC95%: −0,610 a −0,375; p<0,001). El CTG5 fue el alelo polimórfico más frecuente en sanos. Del total de afectos, el 28,3% presentaron la forma leve o asintomática, el 59,1% la forma clásica y el 12,6% la forma severa. El 35,1% presentaron herencia materna, el 59,4% herencia paterna y el 5,5% herencia incierta. En las formas leves la calvicie frontal en varones fue el rasgo fenotípico más prevalente, junto con miotonía y cataratas, mientras que en la clásica predominó la ptosis palpebral, la debilidad facial, las alteraciones en la voz y la pronunciación, la miotonía y la sensación de cansancio/somnolencia.
ConclusionesLa incidencia de DM1 es relevante en Aragón. La revisión multidisciplinar del fenotipo de pacientes con DM1 es clave para un diagnóstico precoz y medicina personalizada.
The incidence of myotonic dystrophy type1 (DM1), a disease with great phenotypic variety, in our region is unknown. This study aims to estimate the incidence of DM1 at our hospital (a reference centre in Aragon, Spain) and to identify the characteristics of our population (genotype-phenotype correlation).
MethodsRetrospective, descriptive study of 459 patients classified according to the number of CTG repeats, as follows: normal (5-35), premutation (36-50), protomutation (51-80), small expansions (81-150), intermediate expansions (151-1000), and large expansions (>1000). Furthermore, according to clinical phenotype, patients were categorised as unaffected (5-50 CTG repeats), mild form or asymptomatic (51-150), classical form (151-1000), and severe form (>1000).
ResultsThe incidence of DM1 was 20.61 cases per million person-years (95%CI: 19.59-21.63). An inverse correlation was observed between the number of CTG repeats and the age at genetic diagnosis (ρ=−0.547; 95%CI: −0.610 to −0.375; P<.001). CTG5 was the most frequent polymorphic allele in healthy individuals. Of all patients with DM1, 28.3% presented the mild or asymptomatic form, 59.1% the classical form, and 12.6% the severe form. Inheritance was maternal in 35.1% of cases, paternal in 59.4%, and uncertain in 5.5%. In mild forms, frontal balding in men was the most prevalent phenotypic trait, as well as myotonia and cataracts, while in the classical form, ptosis, facial weakness, voice and pronunciation alterations, myotonia, and fatigue/sleepiness were most frequent.
ConclusionsThe incidence of DM1 in Aragon is significant. Multidisciplinary study of the phenotype of patients with DM1 is key to early diagnosis and personalised management.
La distrofia miotónica tipo 1 (DM1) o enfermedad de Steinert (OMIM#160900) es una miopatía autosómica dominante (AD) caracterizada por producir una expansión del triplete citosina-timina-guanina (CTG) en una región no codificante presente en el gen DMPK (proteincinasa de la distrofia miotónica) localizado en el brazo largo del cromosoma19 (19q13.3)1-4.
Es una enfermedad que presenta gran variedad fenotípica, y debido a esto, cada paciente precisa tratamiento personalizado5,6. Las manifestaciones clínicas de esta enfermedad estudiadas durante años han sido recientemente recopiladas de forma exhaustiva en la guía clínica publicada en 20195. Entre ellas hay que destacar:
- 1.
Problemas musculares: debilidad de predominio distal, aunque también pueden verse afectados los músculos del cuello, de la cara, de la masticación, de la deglución y de la fonación7.
- 2.
Problemas del sistema nervioso central: manifestaciones muy variables, entre las que se incluyen déficit cognitivo, apatía, fatiga, alteraciones del sueño8 y, en la DM1 de comienzo neonatal, retraso mental, déficit de atención con hiperactividad y dificultad para las funciones ejecutivas9.
- 3.
Afectación cardíaca: entre el 75 y el 80% de los pacientes presentan algún grado de afectación cardíaca, siendo las más frecuentes alteraciones electrocardiográficas y arritmias7,10. El espectro clínico es variable, desde alteraciones leves del electrocardiograma hasta arritmias graves que pueden ocasionar la muerte súbita3,11,12.
- 4.
Afectación respiratoria: suele ser frecuente y constituye una de las principales causas de muerte prematura en estos pacientes. Parece existir una cierta correlación entre el tamaño de la expansión y la intensidad de la afectación respiratoria13-15.
- 5.
Alteraciones dermatológicas: pilomatricomas múltiples, alopecia, dermatitis seborreica, nevus displásico2.
- 6.
Alteraciones endocrinas: hipogonadismo hipergonadotropo. En varones, niveles disminuidos de testosterona junto con elevación de FSH y LH. En mujeres, infertilidad, abortos espontáneos y fallo ovárico precoz16,17.
- 7.
Alteraciones lipídicas: suelen elevarse los niveles de triglicéridos y disminuir los de colesterol HDL; y del metabolismo fosfocálcico: en el 90% de los pacientes existen niveles deficitarios de vitaminaD.
- 8.
Afectación gastrointestinal: disfagia y alteraciones de la masticación por debilidad y miotonía de los músculos masticatorios. En el esófago existe hipotonía, en el estómago, un vaciamiento gástrico enlentecido que se manifiesta con digestiones pesadas y prolongadas; en el intestino, fases de estreñimiento alternadas con diarreas18.
- 9.
Afectación oftalmológica: la mayoría de los pacientes presenta hipotensión ocular y cataratas, que suelen diagnosticarse a partir de los 50años19, junto con ptosis palpebral producida por la debilidad del músculo elevador del párpado.
- 10.
Riesgos en anestesia: la anestesia con opiáceos y sedantes con efecto respiratorio debe utilizarse con cautela, puesto que estos pacientes son especialmente sensibles a estos fármacos20.
Existen algunos estudios sobre la prevalencia de las distrofias miotónicas en diferentes países21 y algunos previos en regiones de España22, si bien en Aragón no hay datado ningún estudio acerca de la incidencia y prevalencia de esta enfermedad fundamentalmente en adultos ni sobre la prevalencia de la gran variedad de manifestaciones clínicas.
El objetivo de este estudio fue evaluar la incidencia de la DM1 en la población de Aragón atendida en la Sección de Genética Clínica del Hospital Universitario Miguel Servet (HUMS) de Zaragoza, centro de referencia en Aragón para la realización del estudio genético de la DM1, entre enero de 2007 y diciembre de 2019 según sexo, sintomatología clínica y edad al diagnóstico genético. Además, se evaluó la prevalencia de determinados rasgos clínicos y se estudió la correlación genotipo-fenotipo. Otros objetivos secundarios fueron analizar el tipo de transmisión paterna/materna, clasificar a los pacientes diagnosticados según el número de repeticiones del triplete CTG en las diferentes subcategorías clínicas, determinar el número de test diagnósticos y predictivos realizados y, finalmente, establecer el número de repeticiones CTG más frecuente en rango normal (alelo polimórfico).
Materiales y métodosSujetos/PacientesEstudio descriptivo retrospectivo que evalúa a 459 pacientes que durante el periodo enero 2007-diciembre 2019 fueron remitidos a la Sección de Genética Clínica del HUMS, hospital terciario, para estudio genético de DM1 (expansión de tripletes CTG del gen DMPK).
Los criterios de inclusión para la realización del estudio genético incluían la presencia de síntomas clínicos o hallazgos electromiográficos compatibles con la enfermedad y/o antecedentes familiares de DM1 que lo justificaran. Se recogieron datos demográficos (sexo, edad de derivación para el estudio genético, lugar de residencia), número de repeticiones CTG, manifestaciones clínicas e historia familiar.
Los pacientes fueron clasificados según el número de repeticiones CTG en las siguientes categorías: normal (5-35CTG), premutado (36-50CTG), protomutado (51-80CTG), pequeñas expansiones (81-150CTG), expansiones intermedias (151-1.000CTG) y grandes expansiones (>1.000CTG). Por otro lado, según el fenotipo mostrado, se clasificaron en: no afectos (5-50CTG), forma leve o asintomática (51-150CTG), forma clásica (151-1.000CTGs) y forma severa (>1.000CTG)2,5,23.
MuestrasEl tipo de muestra utilizado para la realización de los análisis predictivos y sintomáticos fue ADN de sangre periférica con EDTA. En los estudios prenatales se usó ADN de muestra de vellosidad corial.
Métodos/TécnicasPara el análisis genético de DM1 se utilizaron dos técnicas diferentes en el periodo de estudio. Durante los años 2007-2011 la determinación del número de repeticiones CTG se realizó mediante PCR convencional y electroforesis en gel de agarosa seguida de Southern blot en los casos que solo se visualizaba un único alelo. La PCR convencional presentaba la limitación de no poder calcular con exactitud el número de repeticiones del triplete. En el periodo 2012-2019 se realizó el análisis directo de la región (CTG)n inestable del gen DMPK (NM_004409.4) mediante PCR, electroforesis capilar y análisis fluorescente de los fragmentos (Kit Adellgene Myotonic Dystrophy Screening) y Triplet Primed Repeat PCR (TP-PCR) (Kit Adellgene Myotonic Dystrophy Confirmatory) con el secuenciador ABI3130xl y el software Genemapper 4.0. La fiabilidad de los kits es del 99% y la precisión de ±1 repetición en el rango de tamaños ≤50 y ±3 repeticiones en el rango expandido (>51-150CTG). La Sección de Genética Clínica del HUMS participa satisfactoriamente en el Programa de Control Externo de Calidad ofrecido por la Red Europea de Calidad de Genética Molecular (EMQN) desde 2013, garantizando la veracidad de los resultados emitidos con esta técnica.
La incidencia de DM1 se calculó a partir del número de nuevos casos diagnosticados por millón de habitantes por año durante el período 2007-2019. Las estimaciones de población atendida por el hospital en cada año fueron proporcionadas por el Departamento de Gestión de los Sistemas de Información del HUMS.
Análisis estadísticoSe calculó la distribución de frecuencias de cada categoría para cada variable cualitativa. Las variables cuantitativas se exploraron con la prueba de Shapiro-Wilk (bondad de ajuste a una distribución normal) y se calcularon indicadores de tendencia central (media o mediana) y dispersión (desviación estándar o percentiles).
Las diferencias entre grupos según sexo, edad de derivación, resultados del análisis genético y antecedentes familiares se analizaron mediante pruebas de contraste de hipótesis, con comparación de proporciones (χ2) y de medias (t de Student, U de Mann-Whitney) según procediera. El test de correlación de Spearman se usó para verificar la existencia de relación entre el número de repeticiones CTG y la edad de derivación. Todos los datos se analizaron utilizando el software estadístico Jamovi 1.1.9.0 y la significación estadística se determinó en p≤0,05 (dos colas).
Este estudio fue aprobado por el Comité de Ética de la Investigación de la Comunidad de Aragón (CEICA).
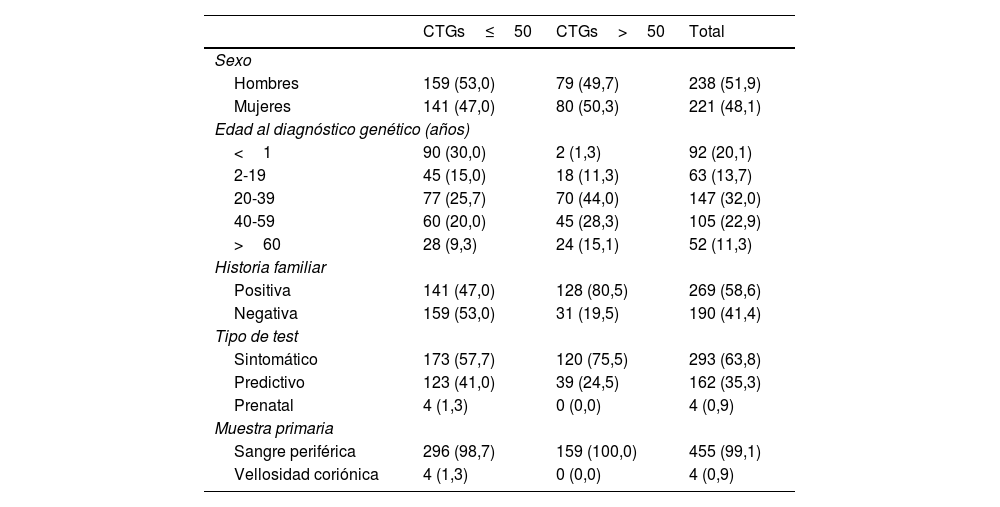
ResultadosSe identificaron 159 (34,6%) casos de DM1 (>50 repeticiones CTG) distribuidos en 73 familias de un total de 459 personas estudiadas. Los 300 (65,4%) individuos restantes se consideraron no afectos (≤50 repeticiones CTG). Los datos demográficos, tipo de prueba, forma clínica e historia familiar de los pacientes de ambos grupos se recogen en la tabla 1.
Datos demográficos, historia familiar y resultados de los estudios genéticos de enfermedad de Steinert
| CTGs≤50 | CTGs>50 | Total | |
|---|---|---|---|
| Sexo | |||
| Hombres | 159 (53,0) | 79 (49,7) | 238 (51,9) |
| Mujeres | 141 (47,0) | 80 (50,3) | 221 (48,1) |
| Edad al diagnóstico genético (años) | |||
| <1 | 90 (30,0) | 2 (1,3) | 92 (20,1) |
| 2-19 | 45 (15,0) | 18 (11,3) | 63 (13,7) |
| 20-39 | 77 (25,7) | 70 (44,0) | 147 (32,0) |
| 40-59 | 60 (20,0) | 45 (28,3) | 105 (22,9) |
| >60 | 28 (9,3) | 24 (15,1) | 52 (11,3) |
| Historia familiar | |||
| Positiva | 141 (47,0) | 128 (80,5) | 269 (58,6) |
| Negativa | 159 (53,0) | 31 (19,5) | 190 (41,4) |
| Tipo de test | |||
| Sintomático | 173 (57,7) | 120 (75,5) | 293 (63,8) |
| Predictivo | 123 (41,0) | 39 (24,5) | 162 (35,3) |
| Prenatal | 4 (1,3) | 0 (0,0) | 4 (0,9) |
| Muestra primaria | |||
| Sangre periférica | 296 (98,7) | 159 (100,0) | 455 (99,1) |
| Vellosidad coriónica | 4 (1,3) | 0 (0,0) | 4 (0,9) |
Expresados en términos de frecuencias absolutas y relativas: n (%).
Entre los casos positivos, la ratio hombre:mujer fue prácticamente 1:1; con 80 (50,3%) mujeres y 79 (49,7%) hombres afectos. Un total de 128/159 (80,5%) casos positivos resultaron tener antecedentes familiares de DM1, bien sospechados clínicamente o confirmados por análisis genético. Entre los casos con historia familiar, 45 (35,1%) presentaron herencia materna, 76 (59,4%) paterna y 7 (5,5%) fueron clasificados como pacientes con herencia incierta al no poder determinarse bien por no disponer del estudio genético de los progenitores realizado en otros laboratorios, por el fallecimiento de los mismos o en casos de reciente diagnóstico en los que aún no se había determinado el patrón de herencia en el momento del estudio.
De todos los estudios genéticos, 293/459 (63,8%) se realizaron en pacientes que presentaban síntomas compatibles con la enfermedad, mientras que 162/459 (35,3%) fueron test predictivos y 4/460 (0,9%) prenatales. Del total de estudios predictivos realizados, 39 resultaron positivos, permitiendo que 39/159 (24,5%) casos de DM1 fueran diagnosticados en ausencia de sintomatología clínica evidente. Se incluyeron además 4estudios prenatales, resultando todos ellos negativos.
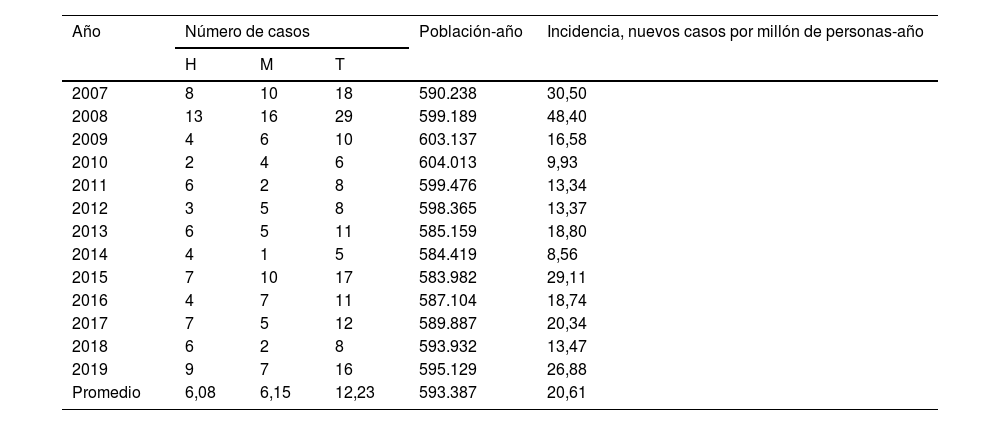
La población total promedio atendida por la Sección de Genética Clínica del HUMS durante el período 2007-2019 fue de 593.387 personas-año, resultando en una incidencia de 20,61 (IC95%: 19,59-21,63) por millón de individuos-año, calculada en base al número de nuevos casos diagnosticados mediante estudios genéticos (tabla 2). Los casos positivos/año para hombres (6,08; IC95%: 5,43-6,73) y mujeres (6,15; IC95%: 5,25-7,05) fueron similares, sin existir diferencias estadísticamente significativas entre ambos (χ2=0,410; p=0,522).
Número de casos e incidencias anuales crudas de enfermedad de Steinert en Aragón, 2007-2019
| Año | Número de casos | Población-año | Incidencia, nuevos casos por millón de personas-año | ||
|---|---|---|---|---|---|
| H | M | T | |||
| 2007 | 8 | 10 | 18 | 590.238 | 30,50 |
| 2008 | 13 | 16 | 29 | 599.189 | 48,40 |
| 2009 | 4 | 6 | 10 | 603.137 | 16,58 |
| 2010 | 2 | 4 | 6 | 604.013 | 9,93 |
| 2011 | 6 | 2 | 8 | 599.476 | 13,34 |
| 2012 | 3 | 5 | 8 | 598.365 | 13,37 |
| 2013 | 6 | 5 | 11 | 585.159 | 18,80 |
| 2014 | 4 | 1 | 5 | 584.419 | 8,56 |
| 2015 | 7 | 10 | 17 | 583.982 | 29,11 |
| 2016 | 4 | 7 | 11 | 587.104 | 18,74 |
| 2017 | 7 | 5 | 12 | 589.887 | 20,34 |
| 2018 | 6 | 2 | 8 | 593.932 | 13,47 |
| 2019 | 9 | 7 | 16 | 595.129 | 26,88 |
| Promedio | 6,08 | 6,15 | 12,23 | 593.387 | 20,61 |
H: hombres; M: mujeres; T: total.
La edad media al diagnóstico genético de los casos positivos fue de 38,9 (DE: 18,6; rango: 0-88) años, siendo el 44,0% de los mismos diagnosticados antes de los 35. No se encontraron diferencias significativas entre la edad media al diagnóstico genético de hombres (40,7; DE: 20,2; rango: 0-88) y mujeres (37,2; DE: 16,8; rango: 0-77) con DM1 (t=−1,19; p=0,234). Para los casos de DM1 con antecedentes familiares negativos, la edad media al diagnóstico genético fue 37,0 (DE: 16,1; rango: 6-63) años, y 39,4 (DE: 19,2; rango: 0-88) para los casos de DM1 con antecedentes familiares, sin existir diferencias significativas (t=−0,638; p=0,524).
El máximo número de repeticiones del alelo expandido encontrado en nuestra población fue 2.800CTG, con un tamaño promedio de 569,9 (DE: 527,7) repeticiones. No se encontraron diferencias significativas entre el número de repeticiones CTG del alelo expandido y el tipo de herencia materna o paterna (U=1.578; p=0,520) ni con el sexo del probando (U=15.386; p=0,216). Por último, la realización de la prueba de Spearman evidenció la existencia de una correlación inversa entre el número de repeticiones CTG y la edad al diagnóstico genético (ρ=−0,547; IC95%: −0,610 a −0,375; p<0,001).
El número mínimo de repeticiones CTG encontrado en la población de estudio fue 5 (CTG5), que además resultó ser el tamaño más frecuentemente hallado en alelos de individuos sanos (20,5%), seguido de CTG11 (9,6%), CTG12 (9,1%) y CTG13 (8,6%). El número de repeticiones CTG promedio hallado en este grupo fue 13,3 (DE: 7,0). En la figura 1 se muestran las frecuencias de repeticiones CTG de los individuos no afectos de nuestra población de estudio.

Del total de casos de DM1, 45/159 (28,3%) presentaron la forma leve o asintomática, 94/159 (59,1%) la forma clásica y 20/159 (12,6%) la forma severa. La distribución por rango de repeticiones CTG y de CTG promedio de cada grupo se muestran en la figura 2.

En relación con el tipo de test genético realizado, 19/45 (42,2%) de los casos con DM1 leve fueron diagnosticados a raíz de presentar síntomas clínicos compatibles con la enfermedad, mientras que los 26/45 (57,8%) restantes fueron diagnosticados mediante test predictivos, en ausencia de sintomatología en el momento de estudio. De los pacientes con DM1 clásica, 84/94 (89,4%) fueron diagnosticados mediante test sintomáticos, mientras que en 10/94 (10,6%) se realizaron test predictivos.
Entre los pacientes con DM1 leve o asintomáticos (81-150CTG) y los protomutados (51-80CTG), el rasgo fenotípico más prevalente fue la calvicie frontal, presente en el 20% de individuos de cada grupo. Las alteraciones en la voz y la pronunciación fueron el segundo hallazgo más frecuente en el grupo de DM1 leve (12%) pero no se encontraron en ningún paciente en el rango de protomutación.
Junto a estas, las alteraciones de la visión, miotonía y sensación de cansancio y somnolencia fueron las características que completaron el cuadro clínico más habitual de los pacientes de estos grupos (20, 36 y 8%, respectivamente, en los individuos del grupo con forma leve o asintomática, y 30, 20 y 20% para los protomutados). Ningún paciente de estos dos grupos presentó alteraciones respiratorias ni diabetes (figs. 3 y 4).
.")
.")
En los pacientes con DM1 clásica (151-1.000CTG) los rasgos más frecuentes fueron la ptosis palpebral (63/94; 67,0%), la debilidad facial (46/94; 48,9%) y las alteraciones de la voz y la pronunciación (44/94; 46,8%) junto con la miotonía (84/94; 89,4%) y la sensación de cansancio o somnolencia (59/94; 62,8%) (fig. 5).
.")
Con respecto a la forma severa (>1.000CTG), las características clínicas se evidenciaron en 17/20 pacientes diagnosticados. Los rasgos más prevalentes fueron la debilidad facial (15/17; 88,2%), la presencia de labio superior en V invertida (15/17; 88,2%) y la ptosis palpebral (14/17; 82,4%). Una vez más, la miotonía fue la manifestación clínica más frecuente (16/17; 94,1%). La presencia de retraso motor, distrés respiratorio y polihidramnios se registró en 5/6 (83,3%) pacientes diagnosticados con menos de 15años de edad y formas congénitas muy severas. Dos pacientes de este grupo fallecieron a causa de la enfermedad, reportando una tasa de letalidad del 10,0% (fig. 6).
.")
Entre todas las familias en estudio destacó la reflejada en la figura 7, en la que se evidenció una herencia paterna (del abuelo, portador de una protomutación, a la madre) y otra materna (de la madre al caso índice), como muestra el pedigrí (fig. 7A). El alelo en rango de protomutación, según estudios recientes23, provoca mayor inestabilidad para la siguiente generación, como pudo observarse en las dos hijas con 500 y 967CTG, respectivamente. Por herencia materna se presentó el caso índice como Steinert congénito, que falleció a los pocos meses de nacer con hipotonía y distrés respiratorio severos. En la figura 7B-E se muestran los electroferogramas obtenidos mediante realización del estudio genético por PCR fluorescente y TP-PCR. Los electroferogramas del test Confirmatory en la protomutación, y especialmente en la forma congénita, evidenciaron una «cola de dragón», confirmándose el alelo expandido con más de 200 repeticiones CTG, cuyo tamaño se determinó exactamente mediante Souther blot.

Árbol genealógico y electroferogramas de casos con protomutación y mutación completa respectivamente. En la parte superior, electroferograma de los test Screening y Confirmatory del abuelo materno. En la parte inferior, electroferogramas de los test Screening y Confirmatory del caso índice.
En nuestro estudio, la incidencia de la enfermedad de Steinert fue de 20,61 casos por millón de individuos-año, pudiéndose considerar relevante. En la literatura, si bien no existen muchos trabajos en los que se calculen tasas de incidencia —a excepción de la de Belgrado (Serbia), de 2,0 casos por millón de individuos-año24, muy inferior a nuestro resultado—, sí se han documentado tasas de prevalencia de DM1 en otras zonas de España (Mallorca: 10,8/100.000 habitantes; región norte: 35,9/100.000 habitantes)22 y en otros países caucásicos, como Italia, Israel, norte del Reino Unido, Serbia y región oeste de Suecia (9,6-11,7, 15,7, 10,4, 5,3, 17,8 casos por 100.000 habitantes, respectivamente)21,25.
El tamaño del alelo polimórfico en rango normal muestra una mayoría de casos con alelos de 5repeticiones CTG seguida de otros de 12CTG. Esto apoya lo descrito previamente, siendo el alelo de 5CTG el mayoritario en otros estudios26,27.
Entre nuestros pacientes afectos de DM1, la ratio hombre:mujer fue prácticamente de 1:1, confirmándose que ambos sexos pueden padecer la enfermedad con la misma probabilidad. Esto contradice a ciertos artículos28 que afirman una afectación mayor en varones que en mujeres.
En la forma leve la principal manifestación clínica fue la miotonía (36% de los afectos). Se observa que esta forma es diagnosticada a edades más avanzadas que la forma clásica o la severa. En general, con más repeticiones del triplete CTG, la clínica será más severa (expresividad variable) y las manifestaciones aparecerán a edades más tempranas (fenómeno de anticipación). La forma leve tiene una franja de expansión del triplete de 81-150CTG, y por ello en esta forma se presenta, además de una miotonía leve, calvicie frontal —principalmente en varones— y cataratas a edades más avanzadas que en los pacientes con formas más severas. En nuestro estudio, a excepción de un caso con 29años y 130 repeticiones CTG, el resto de afectados presentaron cataratas a edades superiores a los 50años.
La forma clásica de la enfermedad es la que más prevalece en este estudio, con el 59,1% del total de casos positivos. Posee multitud de manifestaciones clínicas, sobre las que destaca una miotonía más pronunciada que la de la forma leve, con dificultad para la relajación muscular que se hace patente en un agarrotamiento de manos observable al darle la mano al paciente. La mayoría de los casos poseen un fenotipo facial característico, compuesto por inexpresividad facial (debilidad facial), labio superior en V invertida, ptosis palpebral28, orejas de implantación baja y calvicie frontal (más prevalente en varones). En particular, la prevalencia de ptosis palpebral (67,0%) es similar a la de otros estudios29. Es frecuente la sensación de cansancio permanente y somnolencia/astenia en el 62,8% de los casos, proporción sensiblemente inferior a la de otros trabajos30. Las alteraciones respiratorias son frecuentes —alrededor del 28,0% de nuestros pacientes con DM1 clásica las presentan— y son una de las principales causas de fallecimiento en estos individuos, en los que parece existir cierta correlación entre expansión e intensidad de la afectación respiratoria14. La afectación cardíaca también es frecuente en pacientes con alto número de repeticiones CTG. Las manifestaciones clínicas que suelen darse y que han aparecido en los pacientes de nuestro estudio son arritmias y trastornos de conducción tanto del sistema His-Purkinje como del nodo auriculoventricular31. Pacientes con afectaciones respiratorias y cardíacas poseen expansiones de 500 repeticiones o más, observándose correlación entre un mayor número de repeticiones y una mayor probabilidad de tener afectación en estos órganos32. Por último, cabe destacar que los problemas gastrointestinales se han presentado en una minoría de los pacientes en estudio. Asimismo, se observa una clínica más agresiva conforme aumentan las repeticiones32,33. La afectación gastrointestinal, según diversos estudios2, es una de las más frecuentes y no ha sido bien estudiada, siendo poco valorada tanto por el paciente como por el clínico. En nuestro estudio encontramos un 15,4% de los casos con patología gastrointestinal (∼5% en formas leves o asintomáticas, ∼17,0% en clásicas y ∼28% en severas), pero es muy probable que, en realidad, sean muchos más los pacientes con algún tipo de afectación a este nivel34,35.
La tasa de letalidad fue del 10,0% para las formas severas y/o congénitas, inferior a la de otros estudios36, situada en torno al 16%, aunque en otros llega a encontrarse entre el 16 y el 41%37. Posiblemente el escaso número de pacientes hace que sea poco preciso este porcentaje. Además de las manifestaciones clásicas, en un menor número de pacientes pueden darse problemas a nivel cerebral que provoquen afectación cognitiva. En nuestra población se da en el 19,0% de pacientes con forma severa y en alrededor del 5% con forma clásica. La hipotonía y el distrés respiratorio son los síntomas que más prevalecen en las formas congénitas, al igual que en otros estudios38.
En nuestro trabajo, todos los casos de Steinert severo provienen de herencia materna. En estudios con mayor casuística se han observado casos de Steinert congénito por herencia materna en alrededor del 90%39. No se conoce el mecanismo molecular por el cual la herencia vía materna supone una mayor expansión de CTG y lleva a casos de Steinert congénito. Recientes estudios sugieren la hipótesis de que una mayor proporción de células metiladas provoca esta expansión, aumentando la severidad de la enfermedad33.
Los test predictivos, al hacerse en pacientes asintomáticos o sintomáticos muy leves que se someten al estudio porque tienen algún familiar afecto, suelen realizarse antes de los 30, como ocurre en nuestro estudio, a excepción de dos casos, de 45 y 50años. En algunos de estos se encontraron más de 200CTG, lo que podría explicarse asumiendo que a edades tempranas estos pacientes no habían empezado a manifestar la clínica de la enfermedad.
En cuanto al tipo de herencia, se observó como más prevalente la paterna, con el 59,4%, frente al 35,1% de la materna. Esto puede deberse a que la materna provoca mayor expansión de repeticiones CTG, que conduce a tener descendencia con clínica más agresiva o con Steinert congénito que probablemente no llegue a tener hijos/as. El 5,5% restante poseen un patrón incierto, debido a causas como el reciente diagnóstico de sujetos en los que todavía no se ha realizado el estudio de sus progenitores, pacientes con estudios familiares realizados en otros laboratorios/centros, o pacientes diagnosticados con familiares fallecidos a los que no pudo realizarse el estudio.
Como principal limitación, cabe la posibilidad de que los resultados clínicos de esta serie de casos puedan estar sesgados dada la procedencia de los pacientes, al tratarse de una serie retrospectiva extraída de las bases de datos de estudios genéticos realizados en el HUMS. De esta forma, podrían haberse perdido personas mayores que no se hubieran realizado el estudio genético, personas jóvenes que no quisieron ser estudiadas o personas con cargas sociales o sin descendencia que también rechazaron la posibilidad de realizarse un estudio genético. Estas potenciales pérdidas podrían sesgar los datos analizados.
En conclusión, la incidencia de la enfermedad de Steinert es notable en Aragón. Es necesario la aplicación de nuevas metodologías diagnósticas como la PCR fluorescente y la TP-PCR junto al Southern blot para la confirmación genética de esta enfermedad, obteniendo el número exacto de repeticiones CTG y la realización de una correcta clasificación de los pacientes según correlación genotipo-fenotipo. Se deberían realizar estudios más amplios en los que se evaluaran clínicamente con mayor profundidad alteraciones endocrinológicas, gastrointestinales y otras menos prevalentes. La revisión multidisciplinar de las características clínicas del conjunto de pacientes con DM1 es clave para poder ofrecer un diagnóstico precoz y una medicina personalizada a los mismos. La realización de los estudios genéticos permite detectar a portadores asintomáticos y sintomáticos leves en familiares directos ofreciéndoles un adecuado asesoramiento genético y opciones reproductivas.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.

