




In the ageing process there are some species of non-human primates which can show some of the defining characteristics of the Alzheimer's disease (AD) of man, both in neuropathological changes and cognitive-behavioural symptoms. The study of these species is of prime importance to understand AD and develop therapies to combat this neurodegenerative disease.
DevelopmentIn this second part of the study, these AD features are discussed in the most important non-experimental AD models (Mouse Lemur—Microcebus murinus, Caribbean vervet—Chlorocebus aethiops, and the Rhesus and stump-tailed macaque—Macaca mulatta and M. arctoides) and experimental models (lesional, neurotoxic, pharmacological, immunological, etc.) non-human primates. In all these models cerebral amyloid neuropathology can occur in senility, although with different levels of incidence (100% in vervets; <30% in macaques). The differences between normal and pathological (Alzheimer's) senility in these species are difficult to establish due to the lack of cognitive-behavioural studies in the many groups analysed, as well as the controversy in the results of these studies when they were carried out. However, in some macaques, a correlation between a high degree of functional brain impairment and a large number of neuropathological changes (“possible AD”) has been found.
ConclusionsIn some non-human primates, such as the macaque, the existence of a possible continuum between “normal” ageing process, “normal” ageing with no deep neuropathological and cognitive-behavioural changes, and “pathological ageing” (or “Alzheimer type ageing”), may be considered. In other cases, such as the Caribbean vervet, neuropathological changes are constant and quite marked, but its impact on cognition and behaviour does not seem to be very important. This does assume the possible existence in the human senile physiological regression of a stable phase without dementia even if neuropathological changes appeared.
Existen algunas especies de primates no humanos con algunas de las carac-terísticas definitorias de la enfermedad de Alzheimer (EA) del hombre, tanto en el aspecto neuropatológico como en el cognoscitivo-comportamental, y que son de importancia capital para entender y/o tratar esta enfermedad.
DesarrolloEn esta segunda parte del estudio se analizan estas características durante el proceso de envejecimiento en los modelos de EA más importantes de primates no humanos no experimentales (lémur ratón—Microcebus murinus, cercopiteco verde—Chlorocebus aethiops—y los macacos Rhesus y de cola en tirabuzón—Macaca mulatta y M. arctioides) y experimentales (modelos lesionales, neurotóxicos, farmacológicos, inmunológicos, etc.). En todos estos modelos se puede presentar neuropatología amiloidea cerebral senil, aunque con diferente grado de incidencia (100% en cercopitecos; <30% en macacos). Las diferencias entre senilidad normal y patológica (Alzheimer) en estas especies son difíciles de establecer por la falta de estudios cognoscitivo-comportamentales en muchos grupos analizados, así como por la controversia existente en los resultados de estos estudios cuando se llevaron a cabo. Sin embargo, en algunos macacos se ha comprobado la correlación entre un alto grado de deterioro funcional cerebral y una gran cantidad de alteraciones neuropatológicas (posible «Alzheimer»).
ConclusionesEn los macacos, se puede considerar la existencia de un posible «continuum» entre proceso de envejecimiento «normal», «normal con no profundas alteraciones neuropatoló-gicas y cognoscitivo-comportamentales», y «envejecimiento patológico» o «envejecimiento tipo Alzheimer». En otros casos, como el de los cercopitecos verdes, las alteraciones neuropatológi-cas son constantes y bastante marcadas, pero sus repercusiones cognoscitivo-comportamentales no parecen muy importantes. Ello hace suponer la posible existencia en la involución senil fisiológica de una fase estable sin demencia aun cuando existan alteraciones neuropatológicas.
Certain non-human primates have been thoroughly examined and proposed as animal models for studying cerebral ageing and Alzheimer's disease (AD).1 These animals were chosen for a variety of reasons including their short life expectancy, accumulation of amyloid, and frequent use in laboratories. Each species has its own distinct characteristics with certain similarities and differences with respect to humans with AD; researchers must understand these differences before extrapolating conclusions from animal models to humans. The species whose neuropathological traits have been studied the most belong to the families Cheirogaleidae (dwarf and mouse lemurs) and Cercopithecidae (cercopithecoid monkeys) (see part I).1 Until only recently, the great apes most closely related to humans (especially wild gorillas and captive chimpanzees) were used as subjects in major research projects on behaviour and cerebral function in general. However, such studies have little to do with the question at hand, that is, defining morphological and functional senile involution of the brain in its normal and pathological versions.
Lemurs (prosimians)The species most commonly studied in animal models of AD is Microcebus murinus (grey mouse lemur).2–8 This mammal resembling a rodent is native to Madagascar, and it is now raised in laboratories around the world. Advantages of working with this animal include its small size (body length 12–13cm with a similar tail length; body weight 50–100g) and its short life span (specimens 5 years old are considered elderly). Nevertheless, researchers have reported life spans as long as 14 years in some captive lemur populations. While most lemurs experience slowly progressing cerebral ageing without extreme involution, 20% of the population develops early-onset neurodegeneration. In these primates, neurodegeneration is characterised by massive atrophy of the cortex, hippocampus, basal ganglia, brainstem, and cerebellum. This degeneration is associated with a substantial increase in ventricle size, abundant amyloid plaques, tau protein deposition, and loss of cholinergic neurons. In addition to plaques, subjects present other types of amyloid deposits and argyrophilic neuronal filaments similar to those occurring in many patients with AD. Animals suffering from this form of neurodegeneration lose cognitive and behavioural abilities (the ability to complete tasks and interact socially). These alterations resemble the behavioural changes in human patients with AD. Studies performed to determine the composition, maturation, and distribution of amyloid plaques in Microcebus murinus have also shown certain similarities to human studies. In Microcebus, the β-amyloid deposition pattern may resemble that observed in humans. There are 3 main types of deposits: diffuse plaques measuring some 100 microns in diameter, amyloid plaques with a dense, compact nucleus that are also positive for thioflavin, and loop-shaped formations around blood vessels. The first two formations are also found in patients with AD. As in humans, amyloid deposits in Microcebus have two main components, Abeta42 and Abeta40, that can be revealed using immunocytochemical techniques. Researchers have observed that deposits on cortical vessels were immunopositive for both components, that diffuse deposits were highly positive for Abeta42, and that some plaques were only positive for Abeta42. This suggests that Abeta42 is associated with the early stages of plaque maturation, as has also been observed in humans.9 The ratio of Abeta40 to Abeta42 is higher in Microcebus than in humans, meaning that mechanisms of amyloid production differ somewhat between these species.9 The plaque distribution in Microcebus, listed from high to low density, is as follows: superior part of the temporal lobe, amygdala, prefrontal area, parietal lobe, and occipital lobe. This amyloid distribution pattern is therefore partially similar to the pattern in humans.3,4 Sequencing studies of the gene that codifies APP have revealed that, despite differences in some nucleotides, the Abeta protein and the peptide produced by Abeta catabolism are completely homologous in humans and Microcebus.3 APP in these animals is found in neuron cell bodies and proximal dendrites, in astrocytes and oligodendrocytes, and in amyloid deposits in the cortical parenchyma and vascular walls. This resembles the distribution in humans. Furthermore, researchers have recorded a good correlation between levels of APP and amyloid plaque density, both of which increase with the age of the animal.3,4
Another of the characteristics defining AD in humans, tau protein aggregation, has also been found in Microcebus murinus. The phenomenon increases steadily as the animal ages. The neocortex is the most severely affected area, even in younger individuals. Changes in the subiculum and entorhinal cortex have only been observed in animals older than 8 years (very elderly specimens in this species); this differs greatly from observations in human subjects. Although all animals presenting diffuse beta-amyloidosis also displayed tau protein aggregation in the neocortex, researchers have found no correlations between lesion densities in the different areas that have been studied.3,4
Cercopithecoids (old world monkeys)The Chlorocebus and Macaca genera are the most widely used in research.
The grivet or Chlorocebus aethiops, a species native to Africa but now found in numerous countries in Asia and the Americas, and especially in colonies on the Caribbean island of St. Kitts, was recently proposed as an animal model for amyloid pathology in AD.10–12 Adult male grivets weigh between 5 and 7kg and they can live between 20 and 30 years in captivity (see section 4.1, part I). Studies in individuals aged 15, 22, and 30 years show that at 15 years, the first amyloid deposits (predominantly Abeta 40) begin to form on blood vessels, but that no plaques are visible on the cerebral parenchyma at this stage. Plaques were found on the frontal, temporal, and occipital cortices of all individuals aged 22 or 30 years, and they were more numerous in 30-year-old animals. Immunohistological studies revealed that Abeta 42 was much more abundant than Abeta 40, which is also a feature of the brains of humans with AD. Fibrillary amyloid, which is revealed using thioflavin S staining, was detected over blood vessels in animals of all 3 ages. It was more common in the occipital area, but it was only found in a small percentage of plaques in grivets aged 22 or 30 years; these features differ from findings in humans. Grivets aged 15 years appear to show no neuritic alterations or glial changes, and plaques similar to neuritic plaque only begin to appear in animals aged 22. In contrast, all grivets aged 30 displayed a wide variety of the neuropathological changes typically associated with plaques in human AD (reactive astrocytes, activated microglia, and dystrophic neurites). The large amounts of neuritic plaques at this age, and the fact that plaque density resembled that measured in human AD patients, contrasts with the scarcity of these structures in most non-human primates studied to date.10–12 Nevertheless, although these plaques were immunopositive for various amyloid substances, including APP, only a small percentage tested positive for phosphorylated tau protein, which is a typical indicator of dystrophic neurites in humans. In addition, intraneuronal neurofibrillary tangles also displayed important morphological and histochemical differences with respect to those in humans. This last feature raises important theoretical and practical questions which we will discuss in a later section.
The Macaca genus includes the non-human primates most frequently used in scientific research and featured in the largest number of articles discussing normal and pathological senility in the brain.1Macaca mulatta (Rhesus monkey or macaque) and Macaca fascicularis (also known as M. cynomolgus, cynomolgus, long-tailed macaque or crab-eating macaque) are the most well-known species. The rhesus macaque (Macaca mulatta, Zimmerman, 1780) is native to Asia and its natural range is from Afghanistan to China and Thailand, from sea level to elevations of up to 3000m. They may be found all around the world in captivity or semi-captivity, or as the result of being introduced into new habitats. Until quite recently, captive groups were kept by numerous laboratories. These monkeys measure 45 to 64cm in length and weigh between 5 and 12kg. Social animals, they live in large groups in the wild, and their life span is 35 to 40 years. Nevertheless, individuals older than 20 are considered elderly, and macaques older than 30 exhibit extreme old age.13–15 Differences in longevity between some macaques raised in research centres, wildlife preserves, or protected areas (for examples, the Caribbean keys and small islands) are quite pronounced.10,16 The crab-eating macaque (Macaca fascicularis; Raffles, 1821) is native to Asia (Indochina, Philippines, Indonesia) and its territory extends to altitudes of 2000m. In captivity, its life span can exceed 35 years. This small macaque has a body length of 40 to 65cm and a weight of up to 9kg. It is also known as the long-tailed macaque. In both M. mulatta and M. fascicularis, younger animals (5 years old) lack amyloid deposits in the brain, while many older animals (25–30 years) show deposits in the cerebral cortex.17–19 Nevertheless, the degree of beta-amyloidosis and its functional involvement may be debatable (see section 3). The incidence of tau protein-related intraneuronal neurofibrillary changes is low to non-existent at these young ages,19 but cognitive and behavioural alterations of varying degrees of severity consistently arise in individuals older than 20 or 25. Different authors have described varying types of deficits after employing a range of techniques to evaluate them and correlate them with changes in human normal ageing and AD.19,20 It may be stated that a certain proportion of the total senile age group (constituting a highly variable or imprecise percentage, see section 3) will present more severe morphological and functional changes than those displayed by the rest of the population.13,17–20
APP protein in rhesus macaques is highly homologous to that of humans. A large percentage of the cortical plaques in rhesus macaques are immunoreactive to antibodies selective for human Abeta40, whereas a far smaller percentage is immunoreactive to human anti-Abeta42 antibodies. Fifty percent of the plaques exhibited the enzyme acetylcholinesterase, which had the same biochemical properties as in humans. Another 20% contained apolipoprotein E; a smaller percentage of plaques also contained proteoglycans, heparan sulphate, and α-chymotrypsin. All of the above morphological and histochemical features are also found in AD in humans, but researchers have discovered important differences in the distribution and the spatial and temporal progression of cortical plaques. Specifically, plaque distribution in these animals does not match that in patients with AD, who show more marked plaque deposits in the hippocampus, amygdala, entorhinal cortex, frontal cortex, and temporal and parietal lobes. The classic spatial and temporal disease progression described by Braak and Braak in humans does not appear to occur in these macaque species. However, there are no published systematic studies of this topic that include a sufficiently large colony of observed individuals. Initial deposits form in cortical association areas in rhesus macaques with moderate Abeta levels. In animals with larger quantities of Abeta, these deposits are grouped in paralimbic areas. Areas of the limbic cortex are also affected in macaques with very large amounts of Abeta, but the pattern varies greatly in such cases. Some studies have detected the greatest concentration of plaques in frontal areas and the primary somatosensory cortex, with less dense areas in the amygdala, insula, cingulate cortex, temporal limbic region, occipital cortex, and parietal association cortices.17 The motor cortex and a few sensory areas appear to lack deposits.
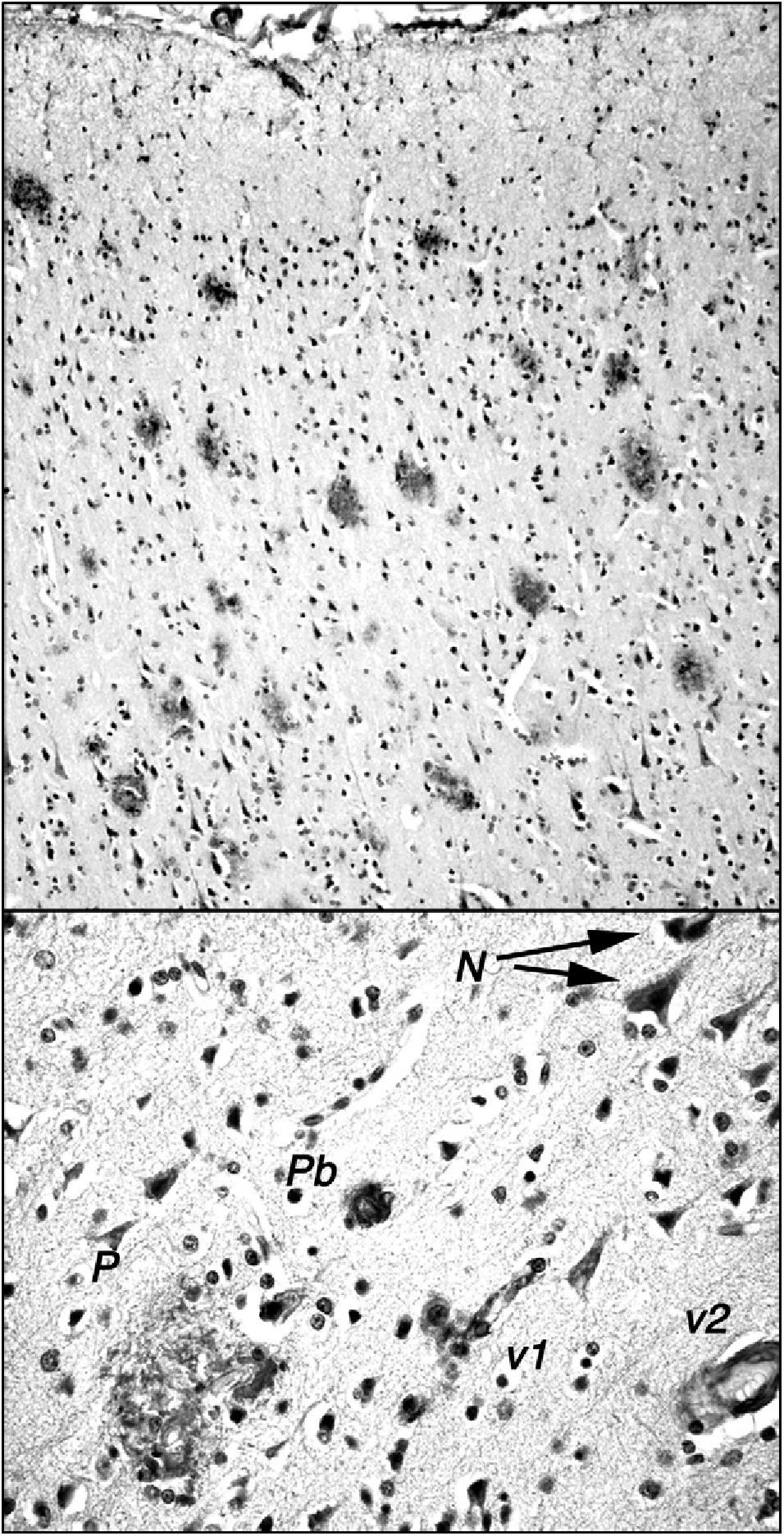
The stump-tailed macaque (Macaca arctoides; G. Saint-Hilarie, 1830) is native to Asia (China, India, Malaysia) and its territory extends to altitudes of 2000m. Colonies are also found on some Caribbean islands, and these macaques have also been raised in captivity or semi-captivity in numerous laboratories in Central America. Their lifespan, given protected conditions, exceeds 30 years. They have a body length of 45 to 70cm and weigh between 7 and 12kg. The stump-tailed macaque is also known as the bear macaque. These macaques demonstrate a morphological and functional involution of the brain similar to that seen in rhesus macaques, but there are a few significant differences. Our research team, which studies a small colony of these macaques in Cuba (Fig. 1), observed that by age 24 to 26, these animals display decreased cognitive and behavioural capacity. This change can be measured using different neuropsychological tests (learning, task performance) and observational study (social interaction, emotivity). Tests that are useful for measuring functional involution and its severity include the delayed matching and non-matching to sample tasks (DMS and DNMS). These tests have been used to show that certain senile individuals in the group had greater behavioural deficits than others. Researchers found neuropathological changes related to amyloid deposits in the post-mortem examinations of all of these animals, which died between the ages of 25 and 34. Areas with a high plaque density were observed in numerous areas of the brain (prefrontal cortex, hippocampus, parahippocampal region). Such areas were present alongside areas without plaque, and scattered plaque also appeared in other regions of the brain (Fig. 2). Plaques are very polymorphous and also present diffuse immunoreactive deposits formed by clusters of small areas showing homogeneous immunostaining or very fine immunoreactive extracellular granulations, as well as some intraneuronal granulations (Fig. 2). These deposits may all be viewed using anti-human Abeta40 and/or Abeta 42 peptide antibodies. However, Abeta40 is the main component, as has also been described in highly evolved simians.21 In some cases, we also observe small intraneuronal accumulations of proteins that react to anti-human tau proteins (phosphorylated and non-phosphorylated). Alongside animals with marked Alzheimer-like neuropathological changes, we find other macaques that exhibit minor cognitive and behavioural changes and very few alterations indicative of neurological disease. In both cases, decreases in neuron count were of little consequence compared to cell density figures from adults younger than 20 years. Only isolated cases of intense gliosis seem to be present in the study population.
 aged 34 years, studied in captivity by the authors at CIREN, the international centre for neurological restoration in Cubanacán, La Habana, Cuba. The individual exhibited marked cognitive and behavioural impairments in the last months of its life; impairment was significantly greater than the mean for its age group. Note the whitish hair typical of old age.")
Stump-tailed macaque (Macaca arctoides) aged 34 years, studied in captivity by the authors at CIREN, the international centre for neurological restoration in Cubanacán, La Habana, Cuba. The individual exhibited marked cognitive and behavioural impairments in the last months of its life; impairment was significantly greater than the mean for its age group. Note the whitish hair typical of old age.
 observed by the International Center of Neurological Restoration (CIREN). This macaque showed cognitive and behavioural deficits that were significantly greater than in ‘normal’ individuals of the same age. Plaques are marked with antibody 6E10 (Chemicon), which recognises epitope 3 to 8 in the human amyloid peptide. The upper section shows the high density of plaques in the frontal cortex; these plaques exhibit little gliosis and few central cores, and they are generally diffuse and non-neuritic. The lower section shows other different types of amyloid deposits: N: small granular intraneuronal accumulations; P: multi-shaped plaque including glial cells and structures with diverse immunoreaction densities; Pb: burnt-out plaque from Alzheimer patients; v1: amyloid reaction adhering to a small blood vessel and surrounding cells; v2: amyloid reaction in the walls of a medium-calibre blood vessel.")
Amyloid plaques in the frontal cortex of a 34-year-old stump-tail macaque (Macaca arctoides) observed by the International Center of Neurological Restoration (CIREN). This macaque showed cognitive and behavioural deficits that were significantly greater than in ‘normal’ individuals of the same age. Plaques are marked with antibody 6E10 (Chemicon), which recognises epitope 3 to 8 in the human amyloid peptide. The upper section shows the high density of plaques in the frontal cortex; these plaques exhibit little gliosis and few central cores, and they are generally diffuse and non-neuritic. The lower section shows other different types of amyloid deposits: N: small granular intraneuronal accumulations; P: multi-shaped plaque including glial cells and structures with diverse immunoreaction densities; Pb: burnt-out plaque from Alzheimer patients; v1: amyloid reaction adhering to a small blood vessel and surrounding cells; v2: amyloid reaction in the walls of a medium-calibre blood vessel.
Published studies have examined the changes arising from manipulation of non-human primate brains in order to produce animals with Alzheimer-like traits (experimental AD models).22,23 Researchers have used a number of methods to produce this effect, including microsurgical or chemical lesions in the cortex or cholinergic nuclei,22–29 and micro-injections of synthetic amyloid or amyloid extracted from the tissues of AD patients.30
The variety of cortical and subcortical lesions in different species have provided considerable advances in our knowledge of cholinergic innervation in the brain22–31 and its potential relationship with the amyloid cascade in AD.24 Nevertheless, we have not yet precisely determined the role played by each of the cortical and subcortical structures, acetylcholine transmitters and receptors in the different behaviours specific to each species. In New World monkeys, lesions to cholinergic nuclei of the basal brain resulted in significantly impaired learning or memorisation ability, but any morphological and functional correlations are difficult to explain. This has been amply demonstrated in marmosets (New World monkey species of different genera within the family Callitrichidae, especially Callithrix, which includes more than 18 species.24,28,29 Likewise, a study in squirrel monkeys (Saimiri sciureus) involved injecting ibotenic acid bilaterally into the nucleus basalis of Meynert. This resulted in cell destruction in the putamen, globus pallidus, striatum, and amygdala, accompanied by deficits in several brain functions including visual discrimination of objects. Other functions, including spatial discrimination, were spared. Nevertheless, the number of damaged cells was not correlated with the quantity of injected neurotoxin or with the degree of loss of capacity. In contrast, lesions in the basal and cortical nuclei in Old World monkeys do not appear to cause significant or lasting memory loss; this is especially true in macaques.26 This may arise because these monkeys have more evolved brains with greater adaptive abilities and more complex neuronal circuits that can set up alternatives when lesions occur (as we also see in humans). It may also be due to technical discrepancies between studies including different primate species (pre-training in different tasks before the lesion, differences on behavioural tests, time between lesion and onset of the study, etc.).8,22,28–30 Regarding differences between Old World monkeys and humans, one might think that impaired brain functions in monkeys might not correspond to those in humans with AD, or that impairment might not be detectable using laboratory tests. In the studies by Ridley et al.,28,29 a period of 2 weeks after surgical intervention was enough time for researchers to observe recovery of certain abilities that primates had lost. The scientists therefore suggested that the modest memory impairment they had observed could be more related to loss of non-cholinergic efferent fibres of the basal brain anterior to the hippocampus and other related limbic system structures than to loss of basal and cortical cholinergic innervation to the cortex.31 The total results from lesion models in non-human primates only add to the controversy regarding the cholinergic theory of AD because the answers they provide are not drawn from the anatomical basis of human cognitive functions.22,23 Furthermore, pharmacological studies in these animals (employing a wide variety of cholinergic agonists and enzymatic regulators) have not produced favourable results that could be used to support trials of any specific AD treatments in humans. In general, treatments shown to improve cholinergic system function in monkeys with lesions in cholinergic basal nuclei have not had positive effects in patients with AD.22,25,29,31
A study published in 1998 indicated that stereotactic microinjections of synthetic Abeta40 delivered to the macaque frontal cortex produced dose-dependent cortical lesions similar to those observed in humans with AD. This was especially marked in elderly macaques. Some time after lesions had been induced, the areas surrounding them presented dystrophic neurons that reacted to silver staining and thioflavin-S staining. Many neurons were immunoreactive to anti-Alz 50 and anti-ubiquitin antibodies.32 Similar studies performed 5 years earlier had failed to demonstrate acute neurotoxicity of amyloid peptides in less-evolved New World monkeys (marmosets of the Callithrix genus).33 However, it was later shown that intracerebral microinjections of liquefied brain tissue from AD patients delivered to middle aged marmosets induced the formation of amyloid plaques, argyrophilic dystrophic neurites, and cerebrovascular amyloid angiopathy.34,35 Beta-amyloidosis had never been observed in marmosets aged 6 to 7. None of these primates exhibited neurofibrillary tangles. We should point out that one of the main objectives of these studies was to clarify whether the beta-amyloidosis or fibrillosis in AD was transmissible, given that prion-related beta-amyloidosis was believed to be transmissible.34,36,37 An intracerebral injection of tissue from a patient with a prion disease was administered to one of these marmosets. The marmoset was later found to have amyloid plaques and angiopathy similar to those observed in brains of monkeys injected with tissue from AD patients.34 These results, obtained using the tools available in the 1990s, supported the idea that some cases of cerebral beta-amyloidosis of any type, including AD, might be induced by exogenous amyloid proteins with beta configuration. These amyloid proteins were infectious even though they lacked nucleic acids, and these cases were considered transmissible diseases.34,35,38 Although AD and prion diseases (transmissible spongiform encephalopathies) have now been placed in separate categories, they do exhibit significant neuropathological and neuropathogenic similarities. The links between them should be used to further our knowledge of such neurodegenerative diseases and determine effective preventive and curative treatments. In the 1990s, Ridley and Baker's group concluded that “beta-amyloidosis may be induced by the introduction of exogenous amyloid beta-protein”.34,35 In 2000, however, they stated that “β-amyloid, or associated factors, can initiate or accelerate the process of cerebral amyloidosis in primates”.36 At present, recently created Alzheimer models in non-human primates feature intracerebral injections of Abeta42 along with substances, such as thiorphan, that prevent it from being mobilised.30 Such models are used both in studies of beta-amyloidosis and in trials of new drugs.
Non-human primates are the animals of choice for trials of potential new drugs or biotechnology products (‘vaccines’) used in AD which may be either preventive or curative. They also feature in animal models studying the possible toxic/neurodegenerative effects of selected substances or environmental conditions and in research into the action mechanisms of neurodegenerative or neuroprotective agents. At the same time, restrictions on the experimental use of primates are increasing, as has been mentioned before.
Regarding use of non-human primates in preclinical trials of AD drugs, animal models have yielded important findings in studies of cholinergic drugs and neurotrophic factors,39–42 and studies monitoring amyloidosis.43,44
Studies of cercopithecoid species have been very important in the development of ‘Alzheimer vaccines’. Macaques injected with amyloid peptides developed high titres of anti-Abeta antibodies with plasma concentrations that were 5 to 10 times higher than in control subjects.45,46 In one study of Chlorocebus aethiops immunised with Abeta peptide over 10 months,47 vaccinated animals had generated large amounts of anti-Abeta antibodies (2–5 times higher than levels in controls) by day 42 of the study. These anti-Abeta antibodies labelled Abeta plaques in humans. Researchers also found antibodies in the cerebrospinal fluid, but at lower titres; they did not observe T-cell response or inflammation. These monkeys did not appear to present significant secondary phenomena at time of immunisation, unlike what occurs in humans. We know that vaccines with Abeta42 began the protocol for the purpose of being marketed at a later time. However, they were discontinued in clinical phase IIa in 2002 since 18 of the vaccinated patients developed meningoencephalitis due to T-cell activation. For this reason, researchers looked for other alternatives for vaccine trials. Later studies employing Abeta15 peptide in macaques showed satisfactory and safe results, including absence of T-cell activation. In the eighth week after vaccination, anti-Abeta42 antibodies began developing in plasma; titres rose with the subsequent inoculations and decreased after the vaccination period. In addition, this antibody was shown to be highly specific against Abeta42.48 The latest results from primate trials show that some researchers are optimistic about the possibility of discovering ‘vaccines’ to prevent or treat AD. It is believed that such drugs may be tested in non-human primates despite the differences in immunological reactions between these species.49 Likewise, results from trials of anti-amyloid monomer or oligomer antibodies in non-human primates seem to admit extrapolation to humans.50
Non-human primates have been employed in studies of the effect of external factors (environment, toxins, living habits) on AD development. We would like to mention the conclusions from 3 lines of research that have been highly influential: restricting caloric intake decreases amyloid accumulation, exposure to certain environmental toxins during childhood may predispose individuals to develop AD, and the consumption of certain metals may elicit neurodegenerative processes.
In the first study, researchers discovered that calorie restriction decreased amyloid deposits in the brains of squirrel monkeys (Saimiri sciureus). This process was shown to be linked to decreased levels of oxidative stress.51 This phenomenon was subsequently demonstrated in macaques.52
In the second study, researchers found that exposure to lead in the early stages of development would induce Alzheimer-like conditions in old age.53 This confirmed earlier findings in rodents that indicated that exposure to lead in early life would predetermine the expression and regulation of the amyloid precursor protein and its amyloidogenic pathway.54 The study was performed in a cohort of Macaca fascicularis females divided into two groups: a control group and an experimental group that received injections of lead acetate during the first 400 days of life. In 2003, the animals were euthanised at the age of 23 and researchers studied a number of their organs, including brains. In elderly animals exposed to lead, researchers found high levels of expression of the genes APP, BACE1 (β-site APP cleaving enzyme 1 or beta secretase), and SP1 (the transcriptional regulator of the other two genes). Levels of Abeta42 were also high (a 100% increase). Regarding typical changes in AD, the brains of animals exposed to lead showed an increase in intracellular Abeta and dense plaques. Researchers also observed diffuse plaques and a few tangles that were morphologically similar to those seen in humans. The activity of DNA methyltransferase 1 (DNMT1) exhibited a 20% decrease, which suggests that the expression of these genes is subject to some type of epigenetic control mediated by mutations. The biomarker for oxidative damage, 8-oxoguanine, was also high. The authors of this study explain this epigenetic phenomenon with the hypothesis that the genes regulated by methylation may be reprogrammed in adulthood. They discovered that 20 out of the 22 genes that were altered as a result of exposure to lead are modifiable by methylation. The idea that humans may develop AD after exposure to lead during their developmental years is supported by the case of a patient who survived severe lead exposure when he was 2 years old but died of severe neurodegeneration at the age of 42 years.55 This patient's brain contained abundant senile plaques and tangles. This study lends weight to the suppositions that numerous environmental agents are involved in the development of AD. It also supports the ‘Barker hypothesis’, which links traumatic experiences or exposure in early stages of life when subjects are particularly vulnerable to adult-onset diseases.56,57
Lastly, several studies have pointed out that certain metals, including manganese and aluminium, may induce neurofibrillary changes.58
DiscussionOur review addresses the question of whether AD exists in primates according to a loose definition of the disease, or if it only occurs in humans. As stated in the introduction, it is impossible for non-human species to experience AD in the strict sense of the term, since no other species possesses the higher functions displayed by the human brain (memory, consciousness, judgement, language, calculation, etc.). In a more general sense, however, it is possible for non-human primates to experience changes in their higher cortical functions as a result of primary degenerative processes in the brain involving a pathogenesis and cognitive and behavioural changes that resemble features of human AD. Numerous authors have concluded that elderly non-human primates (or some of them) do develop marked morphological and behavioural abnormalities similar to those in normal ageing and AD in humans.14,59,60 Nevertheless, this conclusion requires further clarifications regarding not only the type and effects of the abnormalities, but also the percentage of individuals who will present these changes during old age. This section will focus on 3 questions that may be extremely important to furthering our knowledge of AD: (a) the similarities and differences between Alzheimer-like neuropathological lesions in humans and non-human primates; (b) the prevalence of neuropathological lesions and of possible Alzheimer-like cases in non-humans; and (c) involutive-neurodegenerative traits in non-human primate brains and their implications for the way we interpret the relationship between physiologically normal ageing and pathological senile involution (AD).
Similarities and differences between lesions in humans and non-human primatesStudies carried out in a wide variety of non-human primates (from the least evolved species to those most closely related to man) have failed to elucidate why amyloid pathology and tau-related disorders would arise. In fact, these studies have raised question upon question regarding the theories on neurodegeneration and AD. Overall, it is possible to state that elderly primates experience marked neuropathology related to amyloid accumulation, although this does not occur in all species and affects highly variable numbers of individuals within each population. In contrast, the appearance of frank tau-related neuropathology (neurofibrillosis or tauopathy) is exceptional, and it occurs almost exclusively in humans. The following questions also lack answers: why, on the one hand, beta-amyloidosis would always be present in elderly individuals in some species (genus Cercopithecus) but not in others (certain tamarins); and why, on the other hand, beta-amyloidosis would be associated with cerebral neurodegeneration in some individuals of one species (macaques) and absent in individuals with normal ageing, as occurs in humans with AD. It is also difficult to understand the erratic incidence patterns of tau-protein related neurological disorders in the many primate species; numbers are always low except in humans.
APP is the ancestral membrane protein that appears in single-celled organisms. It is also abundantly expressed in mammalian brains. This protein has been conserved with very few variations throughout the evolutionary process; its dual processing pathways, one amyloidogenic and one non-amyloidogenic, exist in all mammals.5,61,62 Nevertheless, beta-amyloidosis does not appear in mammals other than primates (except in very rare cases in bears and dogs which have already been mentioned in the first part of this study).1 The northern tree shrew, Tupaia belangeri, is a proto-primate in the order Scandentia, which is far older than the primate order and closely related to rodents.1 While this tree shrew has a beta amyloid peptide sequence identical to that in humans, studies have not revealed any type of inter-neuronal or vascular beta-amyloidosis.63 In the primate order, amyloid neuropathology seems to present quite clearly and consistently, and it is always related to old age. However, the position of each genus on the evolutionary scale in this order does not appear to correlate to the incidence, prevalence, or intensity of beta-amyloidosis. Similarly, these factors are not correlated with the presence or absence of cognitive and behavioural changes. All of the above lead us to believe that mammals display innate ‘resistance’ to cerebral beta-amyloidosis. This is due to two factors: first, we find one amyloid amino acid sequence in mammals that can experience beta-amyloidosis (primates, dogs, bears) and another in those that do not (rodents, ruminants).36 Secondly, the amino acid sequence in the latter group facilitates the elimination of beta and insoluble products. However, under certain circumstances in old age combined with other unexplained situations, the factors regulating the production and/or elimination of amyloid products become altered in certain primate species, 61 the most evolved mammals. We might suppose that the situations triggering beta-amyloidosis in other primates are those proposed for the development of AD in humans: high levels of oxidative stress, neuroinflammatory reactions, induction of apoptosis, etc.
The morphological and histochemical types of amyloid lesions that present in non-human primates only partially correspond to those described in humans.1,9,17–19,64–66 There are also many varieties of lesions, which is a difficult phenomenon to explain without pointing out that deposits exhibit considerable molecular heterogeneity.67 Section 4.5 described the different types of deposits found in lemurs and macaques: parenchymal plaque, diffuse deposits (non-plaque forming), and perivascular deposits. Deposits in plaque form are more variable in non-human primates than in humans, in terms of both their shape and their immunoreactive characteristics (reactions to Abeta40 and Abeta42 and other components such as neprilysin68). This observation neither corroborates nor invalidates the theories put forth by some authors regarding the origin and progression of these structures in humans. In contrast, diffuse amyloid deposits, whether in the cortex or in subcortical areas, do correlate better with those seen in humans. However, whether in humans or non-human primates, these types of deposits have not been fully studied. Vascular deposits may be very abundant in less-evolved simians when they form strands that surround blood vessels. They are less common in great apes and humans. In addition, as stated before, the progression of amyloid pathology in time and space differs between humans and non-human primates. This is a severe setback for projects considering these primates for Alzheimer models, although in a few cases, researchers have found a good correlation between the amyloid load and brain atrophy.69,70
One of the major inconveniences of using non-human primates in Alzheimer models is the virtual lack of tau-related neuropathology in these species compared to observations of the brains of humans with AD. This means that simians and prosimians show almost no examples of neurofibrillary tangles and dystrophic neurites, and very little neuronal immunoreactivity to use of anti-tau protein antibodies, especially highly phosphorylated tau protein. These data could be interpreted to mean that such forms of beta-amyloidosis in non-human primates would constitute other diseases well-differentiated from human AD. Nevertheless, some of the studies published in recent years seem to contradict this interpretation. Without entering the debate of whether AD is really a form of beta-amyloidosis or a tauopathy (given that both pathological processes occur in AD, although to different extents in different patients), there are two things we should keep in mind. First of all, an entire range of neurodegenerative illnesses or processes (AD, Parkinson's disease, etc.) are currently categorised as tauopathies. In these diseases, tau protein is involved, to whatever extent and in any form (present in soma, dendrites, or axons, where it necessarily induces alterations in neuron structure whether or not it forms deposits). Tau protein is also correlated to other changes specific to each disease, as seen in different cases of AD that may present low or high incidence rates of tau protein abnormalities in neurological disease. Secondly, several studies have used more sensitive techniques (light or electron microscopes and histochemical techniques to achieve greater resolution) to show morphological and histochemical changes related to tau protein (micro-accumulations of more or less phosphorylated deposits in neuronal soma or dystrophic neurites, or accumulations linked to synapse alterations or amyloid deposits.71–73 There is even a published case of a chimpanzee in which researchers detected paired helical filaments.66
Incidence and prevalence of Alzheimer-type neuropathology and possible ‘Alzheimer syndrome’ in different non-human primate speciesAmong non-human primates, some prosimian and simian species demonstrate a fairly constant presence of amyloid deposits among elderly individuals. The clearest example is Chlorocebus aethiops, the vervet monkey; in this species, 100% of aged animals present amyloidosis. Although we lack in-depth cognitive and behavioural studies in vervets, it seems that beta-amyloidosis is not accompanied by severe alterations in brain function that might be interpreted as cerebral neurodegeneration.
Macaques (Macaca mulatta and M. fascicularis), species which are phylogenetically close to the genus Chlorocebus and within the same family (Cercopithecidae), have been used as selective non-human primate models in many studies. Researchers have observed that macaques present generalised mild to moderate cerebral involution in old age; during this stage, only a few individuals present more severe morphological and functional anomalies than the majority of the elderly population.13,17–19 Such individuals may suffer from an Alzheimer-like syndrome. Nevertheless, this conclusion is questionable for several reasons, especially since the prevalence of the syndrome is only poorly defined. This can also be said of the nature and characteristics of cognitive and behavioural changes. The percentage of individuals in this species experiencing the most severe affectation has not been determined even as an estimate, since available studies have low sample sizes. Only one large colony of wild monkeys is under observation. This colony is located on the island of Cayo Santiago, near the coast of Puerto Rico, and it is studied by the Caribbean Primate Research Center, pertaining to the University of Puerto Rico. The colony contains more than a thousand individuals, distributed in numerous family groups,16 but these monkeys have not been used in research projects addressing cerebral involution and neurodegeneration. The findings which the publications mentioned above have provided on this subject are very contradictory. Initially, authors committed to AD studies found very pronounced anomalies in elderly macaques, such as marked neuronal and synaptic loss in the prefrontal cortex and hippocampus, and beta-amyloidosis.9,17–19,74,75 Other studies at a later date found that morphological and histochemical alterations were not so strongly marked, and that neuronal losses were scarce even in the presence of significant cognitive deficits.76–78 These studies even allow us to raise the question of whether AD in humans might be a process partially independent from or preceding beta-amyloidosis.
Very specific signs that may be interpreted as Alzheimer-like syndrome in rhesus macaques include performance on specific cognitive-behavioural tests and the accumulation of different types of amyloid peptides and tau protein (although the latter phenomenon is scarce). Isolated studies of such signs have shown impairment rates of 0% to 100% in different animal study groups.17–19 The highest recorded prevalence rate of amyloid accumulations in elderly rhesus macaques (nearly 100% of the analysed population) was that described by Satu et al. in 2003.17 There were only a few specific cases of elderly macaques with marked amyloid deposits, pronounced changes in the basal nucleus, and severe behaviour and learning disorders (all of which may be indicative of an Alzheimer-like syndrome.13 Studies carried out in other macaques, including the crab-eating macaque or cynomolgus monkey (Macaca fascicularis) and the stump-tailed macaque (Macaca arctoides), provided very similar results. A severe Alzheimer-type amyloidosis appeared in varying percentages (estimated at no greater than 20% or 30% of all elderly individuals). Species-wide prevalence could not be calculated due to the low number of individuals included in studies.17,18 Researchers did find that most animals presented very severe cognitive and behavioural disorders associated with these intense neurological manifestations. In contrast, studies have also found ‘normal’ elderly macaques exhibiting few changes in social behaviour, learning ability, or brain structure.20
Regarding the significance of cognitive-behavioural deficits, researchers express a wide range of opinions about the links between the “higher functions” specific to each species and the neuropsychological and behavioural tests that have been used to evaluate those functions in different species.79,80 When the objective is to study brain structure and function in different species, or overall evolutionary development, these questions are extremely important, but less so for the study of cerebral involution in each species. In the latter case, the more pressing matter is evaluating loss of functions that are vitally important to the individual's survival (learning, strategies for feeding and avoiding harm, etc.) and the individual's continued social relationships.81 All of these functions are very impaired in animals that we might consider to be affected by an Alzheimer-like syndrome.7 A 2007 publication from a symposium on cerebral ageing analysed the characteristics of senile involution in macaques. It concluded that, like humans, macaques display a ‘successful’ and an ‘unsuccessful’ ageing process, depending on the appearance of cognitive and behavioural impairment that is statistically significant compared to the mean for each age group.82
As a whole, the studies point to a possible AD-like syndrome in macaques, but they have been unable to determine its prevalence and similarity to human AD. Meanwhile, studies in vervet monkeys (genus Chlorocebus) seem to indicate an ageing phenomenon that exhibits Alzheimer-like neuropathological changes but which lacks the severe cognitive impairment present in AD. Amyloid accumulation seems to be less neurotoxic when it occurs with high incidence and prevalence rates in elderly animals. Such is the case in C. aethiops, a species in which no severe cognitive and behavioural problems have been described in elderly animals. However, when they occur only in a limited sub-population within a species, as in humans or macaques, that sub-population can be said to experience Alzheimer-like pathological senile involution. In such cases, damage to synapses, neurons, and neural circuits is significant and elicits severe mental and behavioural disorders. The neurotoxic properties of beta-amyloid deposits may differ among primate species including humans, since their composition and macromolecular structures will also be different. The processes by which beta-amyloid monomers and oligomers aggregate to form fibrils and insoluble deposits remain poorly understood, but they can give rise to different pathophysiological types of deposits. One of these types is characteristic (and aetiopathogenic) of AD in humans, while others are not. This view is supported by the fact that some substances react differently to human and non-human amyloid deposits. Especially interesting is the case of the considerable reactivity of PIB (benzothiazole imaging agent Pittsburgh Compound B, a marker for plaques used in PET studies). PIB is seen in humans, but not in other primates (squirrel monkeys, macaques).83
Physiological senile involution and pathological senile involution (AD) The continuum from normal ageing to ADThe extreme variability among Alzheimer-type syndromes in different non-human primate species and different individuals of the same species also has important implications for the development of theories aiming to describe the relationship between physiological ageing and AD. Several studies in the Cercopithecoidea superfamily indicate that there are very different senile involution patterns which may be specific to the genus and species or to individual subjects. Cerebral amyloidosis has a variable degree of involvement in ageing processes, whether those processes are normal, accompanied by mild cognitive/behavioural changes, or typical of pathological neurodegeneration or Alzheimer syndrome. These studies were performed in Chlorocebus aethiops (in which 100% of the animals presented amyloid deposits accompanied by slight behavioural changes) and Macaca mulatta/M. fascicularis (which demonstrated highly variable levels of amyloidosis reaching 100% in certain study groups although levels were at or below 30% for all elderly individuals in these two species; this sub-population included animals with profound cognitive and behavioural anomalies). This leads us to think that we still lack a complete understanding of the cognitive and behavioural implications of cerebral beta-amyloidosis.
Different authors’ views on AD represent two near polar opposites: (a) that AD (sporadic form) is an age-related neurodegenerative disease that is completely distinct from senility and develops in individuals with a genetic predisposition determined by other intrinsic factors (diabetes, vascular problems, etc.) and poorly understood extrinsic factors; and (b) that AD is the last phase in senile involution of the brain. Regarding the latter view, Alzheimer himself believed that the case he described was a manifestation of intense, accelerated cerebral ageing. Many modern epidemiological and evolutionary studies have calculated that AD would be nearly 100% prevalent in a population aged approximately 110 years. The precise genes responsible for human evolution and the development of the most complex regions of the brain are the ones that elicit the onset of AD.83,84 Many experts believe that there is a gradual transition (continuum) from normal physiological ageing to the last phases of Alzheimer, between which we find mild cognitive impairment (MCI). Other experts believe that involution is individualised and results in different situations.85,86 Functional involution of the brain will have unique traits in every individual. Both AD and MCI appear in a wide range of ages from 60 to 100 years, and they present lesion intensities and neuropathological manifestations that do not correlate to the person's age.85–87 Therefore, at specified ages, we find different groups of normal elderly subjects, normal elderly subjects displaying mild neurodegenerative changes, and subjects with MCI/AD with differing degrees of clinical and histological degeneration. These groups may constitute ‘sub-populations’ for various ageing process, or they may represent a single continuous process of cerebral ageing. The latter model would also include very elderly subjects experiencing ‘successful’ ageing from a functional viewpoint, even when examination of their brain tissue reveals neurodegenerative changes.87
Some authors find the following to be conclusive proof of a clear continuum from physiological ageing to AD: results from prevalence studies of AD stratified by age, the finding that one part of the MCI group will develop AD within 4 to 5 years,85,86 and the spatial-temporal progression of AD on both the clinical and pathological levels. These findings, however, may only be indicative of the progressive nature of AD and the fact that human ageing is associated with an increasing propensity for developing the disease. The sub-population of elderly humans without dementia, despite displaying mild to moderate neuropathological changes, could be equivalent to the large sub-population of elderly guenons exhibiting scarce behavioural impairment. This sub-population is of great theoretical interest. On the one hand, this phase could be a starting point for AD-like neurodegenerative disease, which would be a drawback. On the other hand, it could also indicate the presence of a stable, dementia-free phase in normal physiological ageing. Further multidisciplinary studies are needed if we are to stabilise human senile involution in this phase.
Investigating the underlying cause of this special form of senile involution in the human brain will require examining changes that occurred in our evolutionary process.83,84,88–92 Many genes, and many mutations, were selected in the course of evolution that resulted in non-human primates, and finally in humans. Adaptations to an ancestral environment when life expectancy was shorter created a genome that was optimal at that time; at present, given much longer life expectancy, the same genome may be the cause of very significant pathological changes in the final stages of life. This being the case, the evolutionary process that gave rise to the primates seems to have shown some conditions fostering the development of a new age-related degenerative disease that presents its most florid manifestations in humans. Different neuropathological manifestations (amyloidosis, tau-dependent changes, oxidative stress, etc.) among different primate genera and species, including humans, reflect the distinct evolution of APP and tau protein genes with metabolism-related enzymes and mitochondrial respiration enzymes.88–92
ConclusionsConsidered as a group, truly elderly non-human primates differ substantially (both between genera and species and among groups or individuals of the same species) with regard to cognitive/behavioural deficits and in the prevalence, incidence, and type of neuropathological changes (especially amyloid changes) resembling those observed in humans with AD. Neuropathological changes resembling those in human AD mostly involve beta-amyloid; accumulations that are immunoreactive to phosphorylated tau protein only appear in isolated cases. These changes are rarely seen in other mammals, although they may be observed in all primate suborders, from prosimians (lemurs) to great apes (chimpanzees), and also including Platyrrhini (Cebuella, squirrel monkeys) and non-hominoid members of the Catarrhini parvorder (macaques and guenons). Declines in cognitive and behavioural function are not well-documented, but they seem to exist in all truly senile animals that have been studied. However, only certain species exhibit marked degeneration. After analysing published results and examining our observations from a behavioural and neuropathological study of a colony of macaques (Macaca fascicularis, the crab-eating macaque), we conclude that some individuals of certain non-human primate species present a pronounced neurodegenerative syndrome resembling human AD, compared to other age-matched individuals which experience senile involution with few pronounced neuropathological changes and cognitive/behavioural deficits. Nevertheless, this conclusion cannot be extrapolated to all non-human primates. In other species, including some that are closely related in terms of evolutionary development (for example, the grivet or Chlorocebus aethiops, which shares a family with the macaques), neuropathological alterations may appear consistently without there being marked cognitive deficits. Other species may show few alterations of any type. The prevalence of more severe neuropathological changes observed in different species is quite variable, ranging from 100% in Chlorocebus to less than 30% in macaques. The differences between normal and pathological ageing (Alzheimer syndrome) are difficult to establish because of lack of cognitive/behavioural studies in many study groups; there are also discrepancies with regard to method and data interpretation in the few studies that have been carried out. However, in some specific cases, especially in macaques, researchers have confirmed the correlation between substantial functional deterioration of the brain and a high number of neuropathological changes (possible Alzheimer syndrome). In some macaques, researchers observed a possible continuum between the processes of normal ageing, ageing with slight neuropathological and cognitive-behavioural changes, and pathological or Alzheimer-type ageing. In other cases (grivets), neuropathological changes were constant and quite pronounced, although with milder cognitive and behavioural repercussions. This suggests that there may be a stable, dementia-free phase in physiological ageing even when neuropathological changes are present. Studying primary senile diseases in non-human primates is challenging due to the longevity of individuals of certain species and because of the legal obstacles to primate research. Nevertheless, it allows us to reach important conclusions that may be applicable to human AD research, and which may help us explain and interpret AD pathogenesis and discover potential therapeutic targets.
FundingThis study was primarily funded by the laboratories involved; between 2010 and 2011 it also received a National Plan grant (CTQ 2009-09538).
Conflicts of interestThe authors have no conflicts of interest to declare.
Please cite this article as: Toledano A, Álvarez MI, López-Rodríguez AB, Toledano-Díaz A, Fernández-Verdecia CI. ¿Existe la enfermedad de Alzheimer en todos los primates? Patología Alzheimer en primates no humanos y sus implicaciones fisiopatológicas (II). Neurología. 2014;29:42–55.
