



El síndrome de Lennox-Gastaut (SLG) es una de las encefalopatías epilépticas más severas de la infancia, caracterizada por la tríada electroclínica de actividad generalizada de punta onda lenta (POL) en el electroencefalograma (EEG), múltiples tipos de crisis epilépticas y retraso mental. Con este trabajo pretendemos describir el cuadro sindrómico en un paciente con antecedente de encefalopatía hipóxico-isquémica y SLG, y su respuesta al tratamiento con levetiracetam (LEV).
MétodoEstudio descriptivo evolutivo de un niño de 3 años con antecedentes obstétricos de asfixia intrauterina y repercusión multiorgánica, acidosis metabólica, shock hipovolémico y crisis convulsivas con edema cerebral que a los pocos meses de edad desarrolla un síndrome de West, resistente al tratamiento farmacológico. Progresivamente, la semiología de las crisis cambia a episodios de hipertonía generalizada y mioclonías, con actividad electroencefalográfica de punta-onda lenta.
ResultadosCon el diagnóstico de SLG se inicia tratamiento con LEV, observándose una mejoría sustancial en la esfera cognitiva, en el control de las crisis, y en los hallazgos electroencefalográficos.
ConclusionesEl SLG es uno de los síndromes epilépticos más graves en los pacientes pediátricos, tanto por su semiología como por su farmacorresistencia. El levetiracetam puede producir una mejoría cognitiva, además de contribuir al control de las crisis en estos pacientes.
The Lennox-Gastaut syndrome (LGS) is one of the most severe epileptic encephalopathies of childhood, characterized by electro-clinical triad of generalized spike-wave activity, slow (POL) in the electroencephalogram (EEG), multiple types of seizures and development delay. This paper intends to describe the syndrome in a patient with a history of hypoxic-ischaemic encephalopathy and Lennox-Gastaut syndrome, and a good response to treatment with levetiracetam (LEV).
MethodDescriptive study on the development of a 3 year old child with intrauterine asphyxia, multiorgan failure, metabolic acidosis, hypovolemic shock, and seizures with cerebral oedema, who developed a West syndrome, resistant to drug treatment. The semiology of seizures progressively changed to generalized episodes of hypertonia and myoclonus, with slow spike-wave electroencephalographic activity.
ResultsWith the diagnosis of Lennox-Gastaut syndrome the patient was treated with levetiracetam, showing a substantial improvement in the cognitive sphere, in the control of seizures, and electroencephalographic findings.
ConclusionsLennox-Gastaut syndrome is one of the most severe epileptic syndromes in paediatric patients. Levetiracetam can help cognitive improvement, and contribute to seizure control in these patients.
El síndrome de Lennox-Gastaut (SLG) es una entidad constituida por la tríada de los siguientes hallazgos electroclínicos: a) EEG intercrítico con POL generalizada de 1,5 a 2,5Hz en vigilia y paroxismos generalizados de actividad rápida rítmica (PGARR) durante el sueño, b) diferentes tipos de crisis, incluidas crisis tónicas, ausencias atípicas y drop attacks, y c) retraso en el desarrollo mental y/o alteraciones comportamentales. Sin embargo, no existe todavía un consenso sobre cómo denominar a la enfermedad si alguna de las tres características antes descritas no está presente1. En la clasificación de las epilepsias y síndromes epilépticos propuesta por la ILAE en 1989 se incluyó como epilepsia criptogénica o sintomática generalizada y se definió así: «El SLG se manifiesta en niños de 1 a 8 años de edad, pero aparece principalmente en niños preescolares. Los tipos de crisis más frecuentes son axiales tónicas, atónicas y crisis de ausencia, aunque frecuentemente se asocian crisis mioclónicas, tónico-clónico generalizadas o crisis parciales. La frecuencia de las crisis es alta y el estado epiléptico es frecuente. El EEG generalmente tiene una actividad de base anormal, POL<3Hz, y anormalidades multifocales. Durante el sueño, se presentan paroxismos de ritmos rápidos (10Hz). En general, presentan retraso mental. Las crisis son de difícil control y el desarrollo generalmente es desfavorable. En 60% de los casos, el síndrome ocurre en niños que tienen antecedente de encefalopatía previa, pero en otros puede ser primaria»2.
Desde el punto de vista terapéutico, no existe un acuerdo unificado sobre cuáles son los fármacos que consiguen reportar mayor relación eficacia/tolerabilidad en dicho síndrome ya que, por lo general, ninguno de ellos consigue controlar de forma aislada las crisis ni detener el deterioro cognitivo o conductual.
La asociación de ácido valproico con lamotrigina y topiramato constituye el tratamiento de primera elección3-4. La lamotrigina ha demostrado ser de utilidad, en especial en el control de las crisis atónicas con caídas5. El felbamato ha sido efectivo en algunos casos, pues ha demostrado disminuir el número de crisis en un 50%6, pero su uso es limitado actualmente, por su toxicidad. Si bien se ha indicado la utilidad de la carbamacepina y la vigabatrina, su uso puede comportar riesgos, ya que pueden exacerbar las crisis de ausencias o desencadenar estado epiléptico no convulsivo7.
Se han utilizado otras alternativas terapéuticas, como el ACTH y los corticoides, la dieta cetogénica y la inmunoterapia8,9. La callosotomía ha demostrado efectos transitorios en el control de las caídas10.
Describimos el caso de un niño de 3 años con antecedente de encefalopatía hipóxico-isquémica y SLG que presentó una evolución llamativamente favorable tras instaurar tratamiento con levetiracetam.
MétodoSe ha llevado a cabo un estudio descriptivo, longitudinal, de 2 años de duración en un varón de 3 años, conocido en nuestro Hospital desde los 22 meses de edad, que acude a la consulta de neuropediatría para valoración y control evolutivo de una encefalopatía hipóxico-isquémica perinatal. Como antecedentes personales cabe resaltar un embarazo controlado. Cesárea a las 39+2 semanas por sospecha de sufrimiento fetal. Puntuación del test de Apgar 5 (a los 5min de vida), pH intra útero 6,97, pH cordón 6,05. Ingreso en UCI neonatal durante 23 días. Durante el ingreso presenta crisis convulsivas desde las 28h de vida, tónicas de los 4 miembros, por lo que recibe tratamiento con fenitoína y fenobarbital. En la RM cerebral se objetivan hallazgos compatibles con encefalopatía hipóxico-isquémica severa, con afectación de ambos hemisferios y de los haces córtico-espinales acompañado de alteraciones metabólicas en relación con isquemia difusa. Es dado de alta con los diagnósticos de asfixia intrauterina con repercusión multiorgánica: acidosis metabólica, hipoglucemia, shock hipovolémico con cardiopatía isquémica, coagulación intravascular diseminada, hipertensión pulmonar con hemorragia, lesión hepática, hemorragia digestiva, insuficiencia renal y crisis convulsivas con edema cerebral.
Es seguido en la consulta de neuropediatría de su hospital de origen, donde recibe tratamiento con vigabatrina, ácido valproico y lamotrigina, y es diagnosticado de síndrome de West secundario.
A su llegada a nuestro hospital, refiere crisis esporádicas (una al mes) de hipertonía del hemicuerpo derecho, de 1min de duración. Presenta una microcefalia severa y una paraplejía espástica severa, con nula conexión con el observador y ausencia de movimientos voluntarios.
Se realiza un estudio videoelectroencefalográfico de vigilia y sueño espontáneo diurno (con privación de sueño) en el que se objetiva la existencia de una desorganización de la actividad bioeléctrica cerebral de fondo, con un enlentecimiento generalizado de la misma, de gran amplitud, y sobre la que se entremezclaban abundantes anomalías multifocales epileptiformes (paroxismos de ondas agudas, puntas y punta-onda), de forma difusa, aunque de mayor amplitud en áreas frontocentrales. Asimismo, desde el punto de vista clínico, se observan durante el estudio algunos movimientos espasmódicos aislados de tronco y extremidades que aparecen predominantemente en flexión.
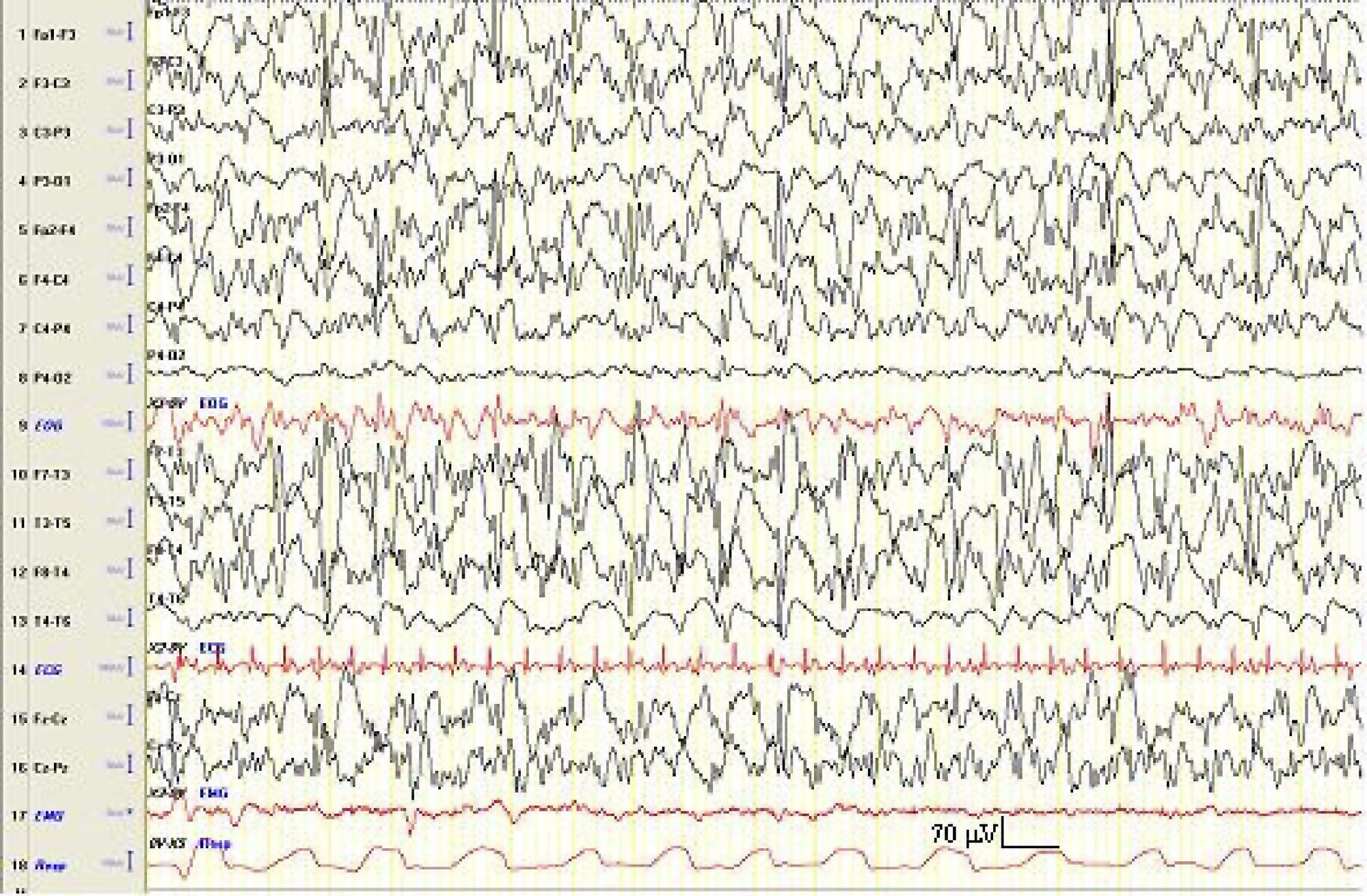
Tras un año sin acudir a las citas, consulta nuevamente por empeoramiento clínico con la aparición de crisis diarias de hipertonía generalizada de 15s de duración, además de otros episodios de desviación de la mirada y la comisura bucal, de segundos de duración. También se observan crisis mioclónicas del hemicuerpo derecho. Se realiza un estudio EEG de vigilia y sueño espontáneo diurno (con privación de sueño) en el que se objetiva la existencia de una desorganización de la actividad bioeléctrica cerebral de fondo, con un enlentecimiento generalizado de la misma, así como la existencia de abundantes anomalías multifocales epileptiformes (paroxismos de ondas agudas, puntas y punta-onda a unos 2Hz), que se presentan de forma difusa, persistente y de mayor amplitud en áreas anteriores (fig. 1).

EEG-poligráfico en el que se objetiva una desorganización de la actividad cerebral de fondo junto con la presencia de paroxismos multifocales/difusos de ondas agudas y punta-onda lenta, de mayor amplitud en regiones anteriores. Constante de tiempo: 0,3s. Filtro de altas frecuencias: 35Hz.
La correlación electroclínica induce a pensar en este momento que se trata de un SLG evolucionado desde un síndrome de West. Se inicia entonces tratamiento con levetiracetam a 35mg/kg, objetivándose una evidente mejoría clínica, con desaparición de las crisis tónicas y mioclónicas, y disminución de las ausencias, además de una clara mejoría en la interacción social.
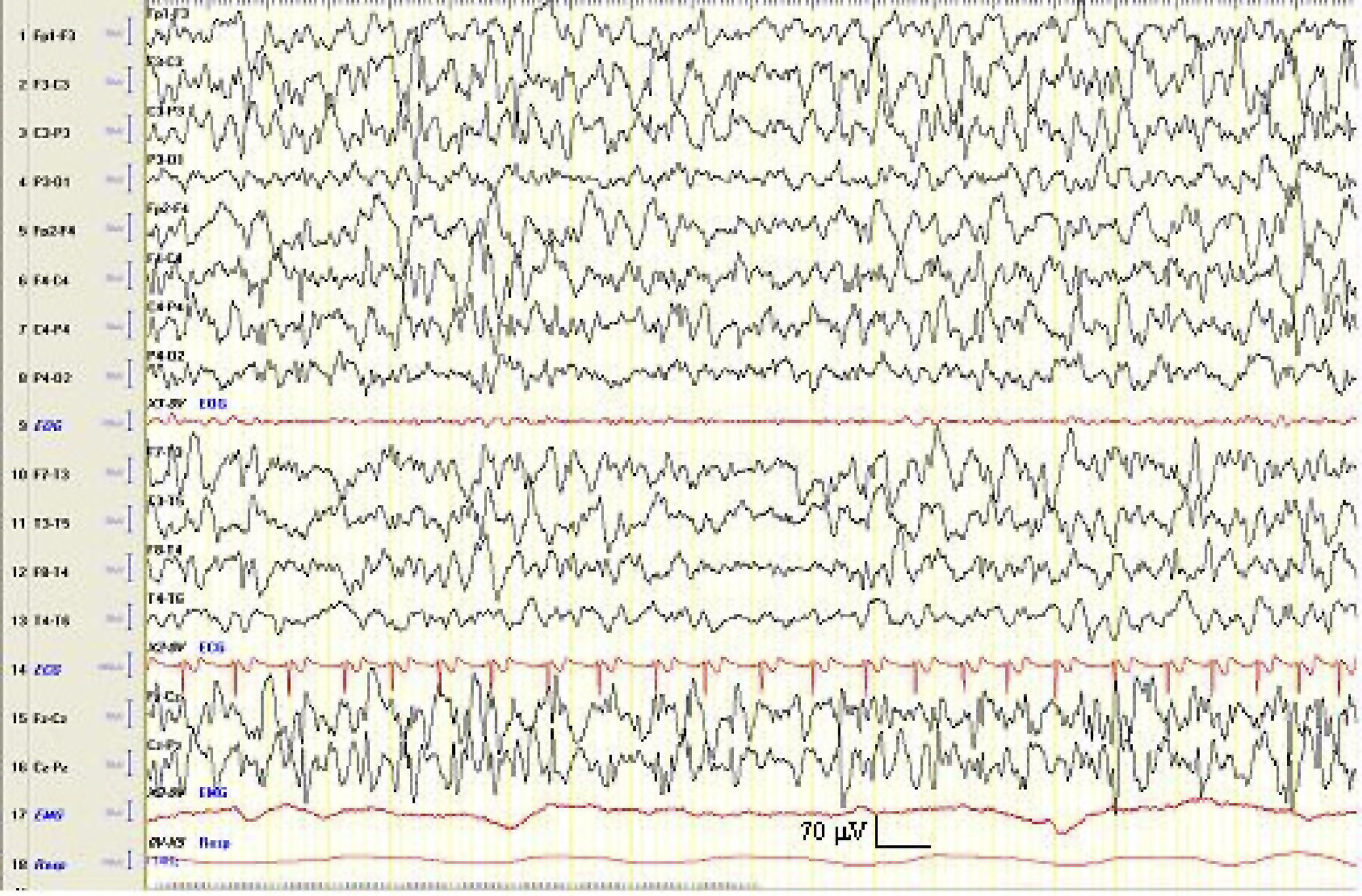
En los sucesivos EEG realizados se aprecia una mejoría de la actividad de fondo, con disminución progresiva en la amplitud y persistencia de las anomalías epileptiformes (fig. 2).
Discusión
El SLG es uno de los síndromes epilépticos descritos más graves en la edad pediátrica11. Está incluido en la clasificación de la ILAE en el grupo de epilepsia y síndromes generalizados de causa sintomática o criptogénica. Su incidencia estimada es de 0,1 por cada 100.000 habitantes, pero su prevalencia es alta (5-10%) debido a su elevada resistencia a los diferentes tratamientos antiepilépticos (representa del 1 al 4% de todas las epilepsias)12,13.
Desde el punto de vista fisiopatológico, la teoría más aceptada es que el SLG resulta de una interrelación compleja de una afección córtico-subcortical multifocal o difusa que ocurre en un tiempo preciso durante el desarrollo madurativo del cerebro1.
El SLG involucra principalmente a los lóbulos frontales, lo cual se refleja en el patrón del deterioro cognoscitivo, que incluye, por ejemplo, el procesamiento rápido de la información y la anticipación; además, la actividad POL predomina en los lóbulos frontales. La edad de inicio del SLG corresponde igualmente con la maduración de las partes anteriores del cerebro, así como con la mielinización de la parte anterior del cuerpo calloso, la cual ocurre en el segundo año de vida permitiendo la sincronización de ambos lóbulos frontales.
Desde el punto de vista etiológico, la forma más frecuente es la sintomática (30-75%), con antecedentes de asfixia perinatal, secuelas de meningoencefalitis, esclerosis tuberosa, displasias corticales, traumatismos craneoencefálicos y menos frecuentemente asociado a tumores o trastornos metabólicos14.
Las manifestaciones clínicas más relevantes para establecer el diagnóstico son15:
- 1.
Crisis tónicas: son las más frecuentes (17-92%), sobre todo durante el sueño lento.
- 2.
Crisis atónicas (26-56%): son la causa responsable más frecuente de caída, pudiendo ser de muy corta duración.
- 3.
Ausencias atípicas (20-65%): con disminución del nivel de conciencia y cuyo inicio y fin son menos bruscos que en la ausencia típica.
Otros tipos de crisis que pueden asociarse son las tónico-clónicas generalizadas, parciales y los espasmos.
En cuanto a la evolución y el pronóstico, debe tenerse presente que se trata de una de las formas más agudas de epilepsia en la infancia, resistente al tratamiento, con evolución frecuente hacia el retraso mental. El peor pronóstico corresponde al grupo del SLG sintomático, especialmente cuando se ha padecido síndrome de West (27-54% de los casos evolucionan hacia un SLG). Los marcadores electroclínicos negativos son: alta frecuencia de crisis, estados epilépticos de repetición y una actividad bioeléctrica cerebral de fondo constantemente enlentecida16.
El tratamiento farmacológico del SLG es complicado y precisa normalmente el uso de politerapia, con resultados en la mayoría de los casos decepcionantes. Se han utilizado diferentes combinaciones de antiepilépticos y existe poca evidencia científica para cada uno de ellos. Los más estudiados son el ácido valproico, felbamato, lamotrigina, topiramato y, más recientemente, la rufinamida. Sobre el levetiracetam no existen estudios controlados que apoyen su utilización, pero sí algunos trabajos que muestran un posible efecto beneficioso en estos pacientes17-19.
En la bibliografía consultada, son llamativamente escasos los trabajos que correlacionan la mejoría clínica (cuando la hay) con la evolución electroencefalográfica favorable.
En un trabajo realizado en el Hospital Infantil de la Universidad de Arkansas se estudió a 6 pacientes con SLG, a cuyo tratamiento se agregó levetiracetam, disminuyendo el número de crisis clínicas así como la actividad electroencefalográfica epileptiforme. Todos los pacientes experimentaron mejoría en el control de las crisis, con especial disminución de las crisis mioclónicas. Es importante señalar la mejoría en el grado de alerta en 3 de estos pacientes, lo cual coincide con lo observado en nuestro caso. El efecto adverso más frecuente fue la irritabilidad, sobre todo al inicio del tratamiento. La dosis de inicio fue de 10mg/kg de peso al día, con una dosis media de 48mg/kg/día19. En nuestro paciente, el efecto beneficioso se observó antes incluso de llegar a esta dosis.
Existen, sin embargo, otros trabajos en los que los resultados son más controvertidos. En 2004 Huber et al20 analizaron la eficacia del LEV en 46 pacientes con epilepsia refractaria y trastornos del aprendizaje de los cuales 7 casos presentaban SLG. El 85% de los casos (6 en total) no respondieron adecuadamente al tratamiento, siendo estos datos negativos de eficacia significativos (p=0,028) frente a epilepsias refractarias de otras etiologías. Asimismo, se objetivó un efecto psicotrópico positivo del fármaco, pero únicamente en los pacientes no afectados por SLG.
En ese mismo año Weber et al21 valoraron de la eficacia del LEV en 10 pacientes con epilepsia generalizada. Entre esos pacientes los autores incluyeron 4 SLG. Los resultados fueron muy dispares; encontraron 2 (50%) casos de mejoría relativa del número de crisis (35 y 40%, respectivamente), 1 caso de empeoramiento (aumento del 25% en número de crisis) y en el último paciente se produjo retirada, nuevamente, por efectos adversos cognitivos. En este estudio los autores no constatan la evolución positiva de la afectación cognitiva sugiriendo, sin embargo, el posible efecto negativo a nivel conductual.
En 2006 Labate et al22 publicaron un estudio observacional de 35 pacientes con diferentes tipos de epilepsia generalizada en las que predominaban las crisis mioclónicas, incluidos 2 pacientes con SLG. En este trabajo los resultados del tratamiento fueron muy poco homogéneos. En un caso se tuvo que suspender por empeoramiento del número de crisis y el otro caso (el de menor edad) experimentó una mejoría de entre el 50-99% en el número de crisis.
No obstante, en todos los trabajos consultados, al igual que en nuestro caso, las crisis mioclónicas fueron las que mejor respondieron al tratamiento con LEV.
A raíz de los resultados obtenidos y tras contrastar los diferentes trabajos publicados sobre el tema en cuestión, podemos señalar que el SLG es uno de los síndromes epilépticos más graves en los pacientes pediátricos, requiriendo frecuentemente el uso de politerapia. El levetiracetam puede producir una mejoría cognitiva, además de contribuir al control de las crisis en estos pacientes, fundamentalmente las mioclónicas. Los hallazgos electroencefalográficos y la correlación electroclínica son herramientas muy valiosas a la hora de predecir la evolución del cuadro y la posible respuesta al tratamiento.
Por último, queremos subrayar la necesidad de más estudios sobre el tema que ayuden a dilucidar las bases terapéuticas de dicho síndrome.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.

