



El síndrome de Brugada es un desorden genético autosómico dominante por mutación del gen SCN5A (causa del 20% de los pacientes con síndrome de Brugada aproximadamente) y SCN1A (causa del 17% del síndrome aproximadamente), que codifica la subunidad alfa de los canales de sodio de la membrana celular1, la cual afecta a la fase 0 y 1 del potencial de despolarización2; al ser esta una canalopatía se ha asociado en algunos casos a crisis epilépticas. Presentamos a continuación 2 casos de pacientes con síndrome de Brugada y epilepsia asociada, posteriormente revisamos la bibliografía sobre esta peculiar asociación.
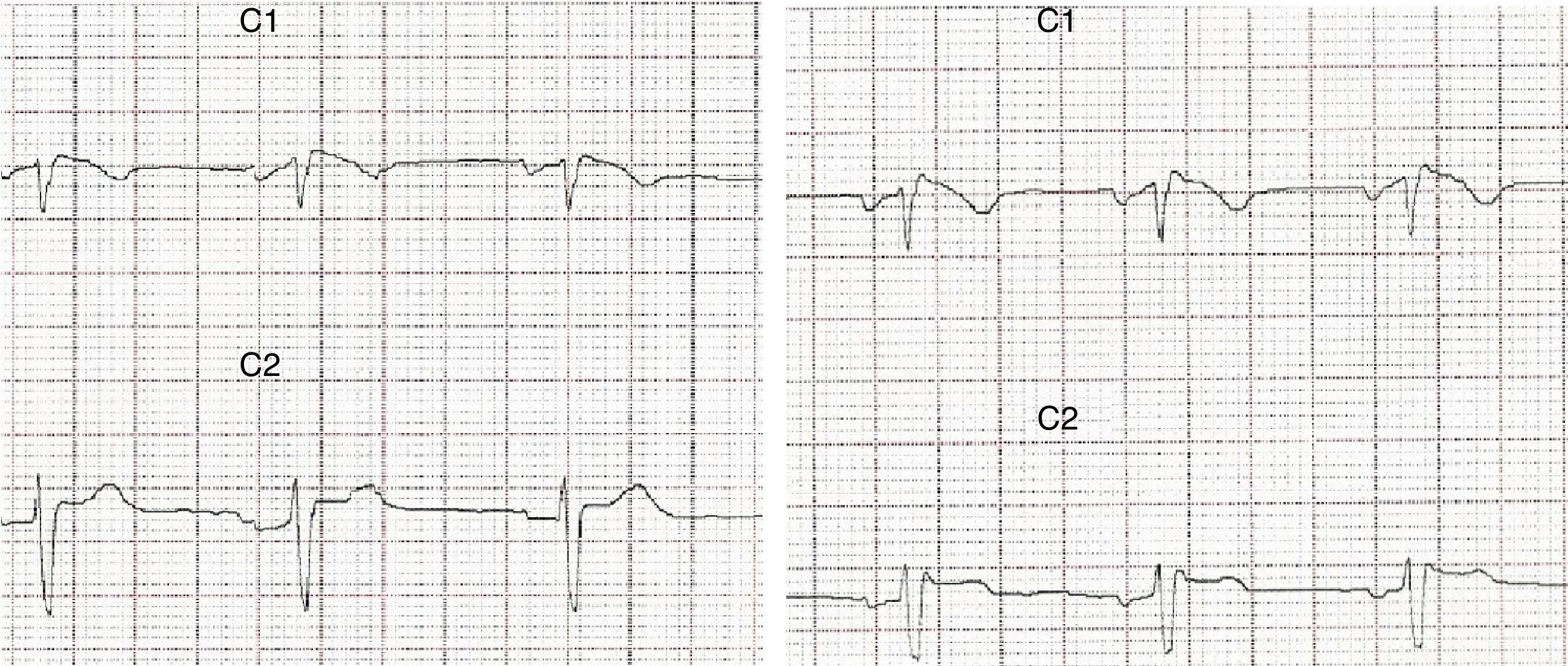
Caso 1Paciente de 37 años, con antecedentes familiares de padre con marcapasos por arritmia cardiaca desconocida, antecedentes personales de síncopes a repetición, acude a urgencias por crisis tónico clónicas autolimitadas con estado confusional posterior. Analíticas sanguíneas normales. TC cerebral y RMN sin alteraciones, el electrocardiograma (ECG) mostraba rSr¿ en V1 con inversión de onda T y V2 con leve elevación de segmento ST (fig. 1), Holter ECG mostraba ritmo sinusal con extrasistolia auricular frecuente, test de ajmalina positivo compatible con síndrome de Brugada, ecocardiograma normal, se implantó desfibrilador automático implantable. Las crisis se controlaron completamente con ácido valproico 1.500mg/día, estando el paciente libre de crisis hasta el día de hoy.
Caso 2
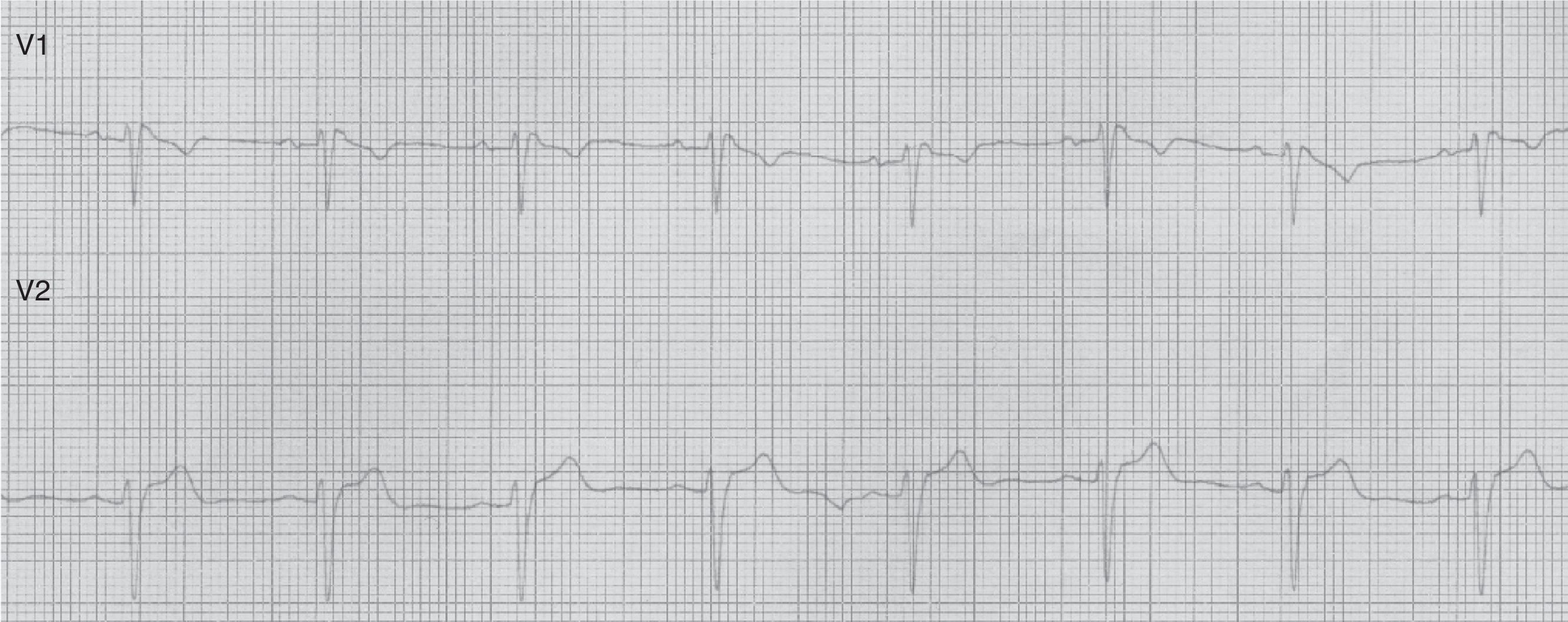
Paciente de 49 años de edad, con antecedentes de hipertensión arterial. Antecedentes familiares de muerte súbita paterna a los 57 años y convulsiones febriles en familiar de primer grado. Ingresado por presentar 2 episodios, mientras dormía, de convulsiones tónico clónica de minutos de duración de la que se recuperó espontáneamente, presentando confusión posterior. Examen físico estrictamente normal. En el ECG practicado se observó taquicardia sinusal, complejo QRS con patrón rSr’ en V1 y segmento ST cóncavo hacia arriba y mínimamente supradesnivelado en V2 (compatible con patrón Brugada tipo 3) (fig. 2).

La RMN cerebral normal, analítica sanguínea normal, Holter normal, ecocardiograma normal, EEG un foco de puntas fronto-temporal derecho más expresivo en fase 3-4. Test de flecainida positivo. Se inicia tratamiento con levetiracetam 2.000mg/día con lo cual paciente está libre de crisis.
El síndrome de Brugada se caracteriza por alteraciones en el ECG, que predisponen taquiarritmias y muerte súbita, la misma suele ocurrir alrededor de los 40 años de edad, típicamente se presenta cuando el paciente está en reposo y por la noche, se presume que uno de los factores precipitantes sería el aumento del tono vagal o estados febriles3. El patrón electrocardiográfico de Brugada tipo I es el bloqueo de rama derecha incompleto (rSR), elevación del segmento ST más de 2mm en las derivaciones V1 a V3 y ondas T invertidas, aunque también se han descrito en las derivaciones inferiores4; el patrón electrocardiográfico silla de montar (rSr’ con el brazo descendente de r’ coincidiendo con el inicio del segmento ST) corresponde al tipo II y III; dado que estos cambios electrocardiográficos son transitorios, aleatorios y pueden no estar presentes en un ECG rutinario, se puede utilizar bloqueadores de los canales de sodio como procainamida, ajmalina o flecainida para desencadenar los cambios típicos en el ECG y llegar así al diagnóstico. El gen SCN5A también esta descrito como causa de síndrome de QT largo en función de si la mutación provoca una pérdida o aumento de la función de la subunidad del canal de sodio5 (en el caso de síndrome de Brugada existe una disminución de su función).
Las canalopatías hereditarias son asociadas a disfunción paroxística de tejidos excitables (corazón, cerebro, músculos). Existen modelos experimentales en ratones donde se encuentra que la proteína del gen SCN5A se expresa en el lóbulo límbico y que podrían tener una función relevante al momento de la activación neuronal6; además este gen está particularmente activo en los astrocitos, lo cual significa que cualquier alteración en el mismo provocaría una susceptibilidad a actividad convulsiva recurrente7. Es de resaltar que en el caso clínico 2, la clínica y los hallazgos podrían estar relacionados con epilepsia nocturna del lóbulo frontal, la cual tiene un componente autosómico dominante asociado a alteraciones de canales iónicos, es así que en la bibliografía encontramos de que si se trata de un paciente sano con convulsiones nocturnas e incontinencia urinaria deberíamos valorar la posibilidad de síndrome de Brugada8.
Los defectos moleculares de los canales de sodio pueden estar sensibilizados a la temperatura, es así que se reportó un caso de fiebre y alteraciones de conducción cardiaca en 2 pacientes9,10.
Entre otras canalopatías con epilepsia y afección cardiaca se encuentran también las canalopatías de potasio11: es así que la afección del canal KCNH2 provoca síndrome QT largo tipo 2 y KCNQ1, que es la causa de síndrome QT largo tipo 1.
El reconocimiento de alteraciones de la conducción cardiaca en pacientes epilépticos es no solo difícil sino también pobre, y lo mismo ocurre en el caso contrario. Una de las pistas que nos puede aportar al reconocimiento de canalopatías de sodio es el empeoramiento de la sintomatología y los cambios en el ECG a la administración de antiepilépticos cuyo mecanismo de acción sea el bloqueo de los canales de sodio.
FinanciaciónDeclaramos no haber recibido ningún tipo de financiación pública y/o privada para la realización del presente trabajo.
El presente trabajo no fue presentado anteriormente en ningún medio escrito, ni en congresos y/o reuniones científicas.

