





To describe the epidemiological and clinical-electroencephalographic characteristics, and associated morbidity of patients with hypothalamic hamartoma, as well as the treatment followed and outcomes.
Patients and methodsWe have retrospectively reviewed the medical histories of 10 patients diagnosed with hypothalamic hamartoma by magnetic resonance imaging (MRI) over the last 20 years.
ResultsThe age of onset of epilepsy in patients with hypothalamic hamartoma in our series was between the first days of life and 2 years. Of the 10 total patients, 8 had epileptic seizures during its progress. All of them had gelastic seizures, in addition to other types of seizures, with the most common being partial simple seizures. The electroencephalographic findings recorded were highly variable. One of the patients developed epileptic encephalopathy. Five patients had some kind of conduct disorder. Five patients had cognitive problems. At least 2 different antiepileptic drugs (AEDs) were measured in 8 of the patients who had seizures, and in 6 of these some type of non-pharmacological treatment had been used with the objective of seizure control. Acceptable control over epilepsy has only been achieved in 3 of the 8 patients. Five patients of the series developed precocious puberty. The average time of follow-up of the series was approximately 6 years.
ConclusionsEpilepsy is the most frequent manifestation of hypothalamic hamartomas. Most cases were drug-resistant, which led to difficulties in the management of these patients, requiring surgery for their control on many occasions. Psychiatric comorbidity and cognitive impairment are common.
Describir las características epidemiológicas, clínico-electroencefalográficas y la morbilidad asociada de los pacientes con hamartoma hipotalámico, así como la evolución y el tratamiento seguido.
Pacientes y métodosSe han revisado retrospectivamente las historias clínicas de 10 pacientes diagnosticados de hamartoma hipotalámico por resonancia magnética en los últimos 20 años.
ResultadosLa edad de debut de la epilepsia en los pacientes con hamartoma hipotalámico en nuestra serie está comprendida entre los primeros días de vida y los 2 años. De los 10 pacientes totales, 8 tuvieron crisis epilépticas en su evolución. Todos ellos presentaron crisis gelásticas, además de otros tipos de crisis, siendo las más frecuentes las parciales simples. Los hallazgos electroencefalográficos registrados fueron muy variables. Uno de los pacientes desarrolló encefalopatía epiléptica. Cinco pacientes presentaron algún tipo de trastorno de conducta. Cinco pacientes presentaron problemas cognitivos. En los 8 pacientes que presentaron crisis se ensayaron al menos 2 fármacos antiepilépticos diferentes y en 6 pacientes de estos se recurrió a alguna modalidad de tratamiento no farmacológica con el objetivo del control de las crisis. Solo en 3 de los 8 pacientes se ha conseguido aceptable control de su epilepsia. Cinco pacientes de la serie desarrollaron pubertad precoz. El tiempo medio de seguimiento de la serie es de 6 años.
ConclusionesLa epilepsia es la manifestación más frecuente de los hamartomas hipotalámicos, siendo en la mayoría de los casos farmacorresistente, lo que conlleva dificultades en el manejo de estos pacientes, precisando en muchas ocasiones cirugía para su control. Es frecuente la aparición de comorbilidad psiquiátrica y afectación cognitiva.
Hypothalamic hamartoma is a non-neoplastic malformation that appears in the hypothalamus between the infundibular recess and the mammillary bodies. It is associated with endocrine and neurological symptoms. The prevalence of this tumour in children and adolescents is approximately 1–2 cases/100000 inhabitants.1 In most cases, it is a sporadic tumour, but in rare cases it may be associated with Pallister-Hall syndrome, an autosomal dominant disorder which includes additional congenital malformations such as polydactyly, imperforate anus and spina bifida or bifid uvula.2
One of the main characteristics of hamartoma is its intrinsically epileptogenic activity, due to the presence of clusters of small GABAergic interneurons which fire spontaneously.3–6
Gelastic seizures are one of the most characteristic and frequent symptoms in patients with hypothalamic hamartoma. These seizures appear in the early years of life (some cases have even been described in neonates), with brief, stereotypical and frequent episodes (sometimes in clusters) of unprovoked and automatic laughter, without any sense of joy or loss of consciousness, although there may be a brief decrease in consciousness. It is usually accompanied by autonomic signs (tachycardia, breathing disorders, flushing, pupil dilation, etc.).7–9 Some patients experience gelastic and dacrystic (crying) seizures at the same time. Patients having these seizures may exhibit groaning and flushing, followed soon after by crying. This may be accompanied by orofacial automatisms.10 There are several descriptions of patients with status gelasticus in the literature.11–14
Patients with hypothalamic hamartoma may suffer other kinds of epileptic manifestations, such as complex partial and generalised seizures.9,12–14 Their development is attributed to a process of secondary epileptogenesis.15
The surface electroencephalogram (EEG) has a limited ability to show epileptiform activity in this pathology, given the deep location of this lesion and the complex connections of the hamartoma.16 During the early stages of the disease, intercritical EEG findings are usually normal and gelastic seizures show a diffuse depression of background activity.17,18
The clinical spectrum of hypothalamic hamartoma is quite variable; patients may have asymptomatic tumours, isolated endocrine disorders such as precocious puberty, or suffer from the syndrome described by Berkovic in 1998 as early-onset gelastic epilepsy and hypothalamic hamartoma (precocious puberty). This syndrome is characterised by catastrophic epileptic encephalopathy and accompanied by cognitive problems and severe behavioural disorders.19–22
Epilepsy associated with hypothalamic hamartoma is typically refractory to AEDs. Achieving good seizure control is rare, even when administering high doses of AEDs in polytherapy.1,11
It has been shown that hamartoma resection is one of the best options for controlling seizures, since they are known to be resistant to AEDs. In addition, this approach produces improvements in cognitive problems and behavioural disorders.23,24
Several surgical approaches have been proposed for resecting hamartomas (microsurgical resection or disconnection, endoscopic resection). However, all those procedures entail substantial surgical risks. For that reason, unconventional surgical procedures delivering acceptable outcomes have been developed recently (gamma-knife radiosurgery, radioactive seed implants, etc.).25,26
Patients and methodsWe reviewed the clinical histories of the patients recorded in our databases as being diagnosed with hypothalamic hamartoma in the last 20 years (between 1990 and 2010).
We obtained epidemiological data (age, sex, race, pregnancies, childbirth, neonatal period, family history), clinical data (age at diagnosis, symptoms, diagnostic delay, associated comorbidity), and complementary tests (EEG, video-EEG, brain magnetic resonance imaging), neuropsychological assessment, and any treatments received.
In cases requiring surgery, we collected data regarding the type of surgery, age at surgery, and any surgical complications.
All patients in our group were paediatric patients (age 0–12 years) at time of diagnosis. They received medical follow-up in our department for 6 years.
All patients were treated in our paediatric neurology, neurosurgery, and endocrinology departments, as needed. No patients were lost to follow-up.
The 5 patients who underwent neuropsychological assessment completed age-adapted cognitive and language tests.
ResultsEpidemiological and perinatal data (Table 1)Of the 10 patients in our series, 6 were male. Only one patient had a family history of epilepsy. Regarding personal medical history, the prenatal period was monitored in all but one of the patients. In another patient, the third-trimester ultrasound showed an intracranial mass anterior to the cerebellum and inferior to the thalamus and third ventricle. Two patients needed to be hospitalised upon birth. One had paroxysmal attacks (the patient mentioned above who was diagnosed prenatally by ultrasound); the other required ventriculoperitoneal shunting on the 6th day of life due to hydrocephalus secondary to an arachnoid cyst.
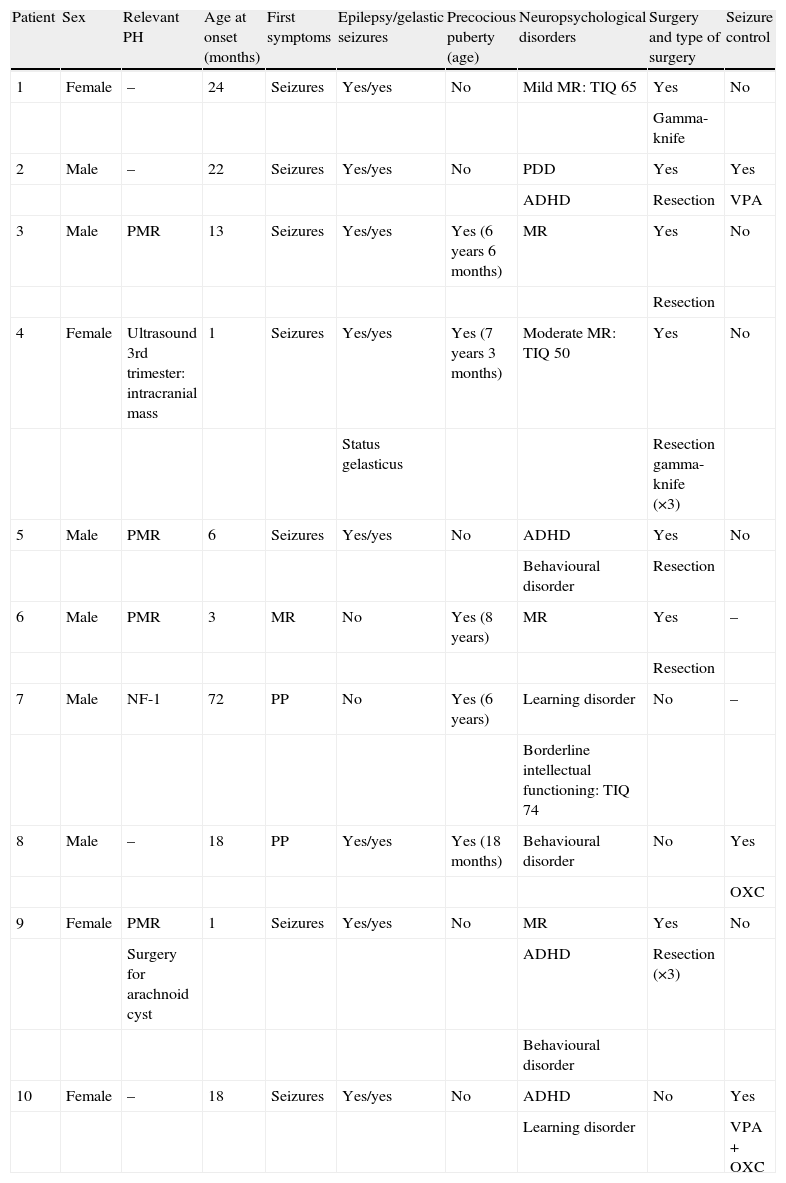
Summary of the characteristics of patients with hypothalamic hamartoma in our series.
| Patient | Sex | Relevant PH | Age at onset (months) | First symptoms | Epilepsy/gelastic seizures | Precocious puberty (age) | Neuropsychological disorders | Surgery and type of surgery | Seizure control |
| 1 | Female | – | 24 | Seizures | Yes/yes | No | Mild MR: TIQ 65 | Yes | No |
| Gamma-knife | |||||||||
| 2 | Male | – | 22 | Seizures | Yes/yes | No | PDD | Yes | Yes |
| ADHD | Resection | VPA | |||||||
| 3 | Male | PMR | 13 | Seizures | Yes/yes | Yes (6 years 6 months) | MR | Yes | No |
| Resection | |||||||||
| 4 | Female | Ultrasound 3rd trimester: intracranial mass | 1 | Seizures | Yes/yes | Yes (7 years 3 months) | Moderate MR: TIQ 50 | Yes | No |
| Status gelasticus | Resection gamma-knife (×3) | ||||||||
| 5 | Male | PMR | 6 | Seizures | Yes/yes | No | ADHD | Yes | No |
| Behavioural disorder | Resection | ||||||||
| 6 | Male | PMR | 3 | MR | No | Yes (8 years) | MR | Yes | – |
| Resection | |||||||||
| 7 | Male | NF-1 | 72 | PP | No | Yes (6 years) | Learning disorder | No | – |
| Borderline intellectual functioning: TIQ 74 | |||||||||
| 8 | Male | – | 18 | PP | Yes/yes | Yes (18 months) | Behavioural disorder | No | Yes |
| OXC | |||||||||
| 9 | Female | PMR | 1 | Seizures | Yes/yes | No | MR | Yes | No |
| Surgery for arachnoid cyst | ADHD | Resection (×3) | |||||||
| Behavioural disorder | |||||||||
| 10 | Female | – | 18 | Seizures | Yes/yes | No | ADHD | No | Yes |
| Learning disorder | VPA+OXC |
PH, personal history; OXC, oxcarbazepine; PP, precocious puberty; MR, mental retardation; PMR, psychomotor retardation; ADHD, attention deficit hyperactivity disorder; PDD, pervasive developmental disorder; VPA, valproic acid.
From the earliest stages, 4 patients experienced psychomotor retardation. One female patient had a previous diagnosis of neurofibromatosis type 1.
Types of presentation (Table 1)Age at symptom onset was highly variable, ranging from the first days of life in 2 patients in the series, to 6 years in the patient who had a hamartoma that was incidentally discovered while using MRI to assess neurofibromatosis type 1.
Age at referral, whether for purposes of beginning the study or for follow-up in the neurology department, also varied (2–14 years). Mean follow-up time in the series was 6 years. Three patients were transitioned from our department after reaching the age of 18.
The initial clinical symptoms of patients with hypothalamic hamartoma were as follows: epileptic seizures in 7 patients, precocious puberty in 2, and psychomotor retardation in the last patient. Of the series total, 2 patients did not experience any seizures during the course of the disease. Age at onset of seizures ranged between a few days and 4 years, with a mean age of 10 months.
Gelastic seizures were common in all patients suffering from epilepsy, and were the first type of seizures experienced by 5 of the patients. Seizures appeared during the first days of life in 2 patients. One patient presented status gelasticus at the age of 11. Dacrystic seizures also appeared in 2 patients; the initial critical episode in 1 of these patients was dacrystic. All patients in the series experienced at least one other type of seizure as well. Simple partial seizures were the most frequent, followed by complex partial and generalised seizures. One of the patients had atonic seizures.
Progression (Table 1)Five patients developed precocious puberty during the course of the disease. All but one was male. Among these patients, 2 had growth hormone deficiency and one also suffered from hyperthyroidism.
In this series, 6 patients had some type of developmental delay or learning disorder. One patient's condition progressed to pervasive developmental disorder. We performed a neuropsychological assessment of 5 patients. The intelligence quotient was within the normal range in 2 patients, with borderline intellectual functioning in 1 patient, mild mental retardation in 1 patient and moderate mental retardation in the last patient.
Five patients had behavioural disorders: there were 2 cases of attention deficit and/or hyperactivity disorder and 2 cases of aggressive conduct. One patient presented both disorders.
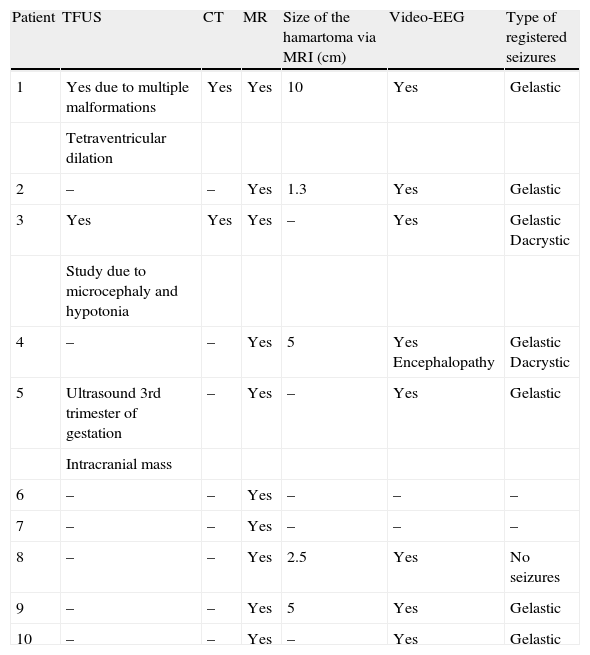
Complementary tests (Table 2)Different imaging tests were performed, including transfontanellar ultrasound in 3 patients due to different reasons. Computed tomography (CT) was carried out in only one patient. The presence of hypothalamic hamartoma was confirmed in all patients by using pituitary MRI (Figs. 1 and 2). The tumour sizes measured by MRI ranged from 1.3cm to 10cm (Table 2).


Summary of complementary tests and findings.
| Patient | TFUS | CT | MR | Size of the hamartoma via MRI (cm) | Video-EEG | Type of registered seizures |
| 1 | Yes due to multiple malformations | Yes | Yes | 10 | Yes | Gelastic |
| Tetraventricular dilation | ||||||
| 2 | – | – | Yes | 1.3 | Yes | Gelastic |
| 3 | Yes | Yes | Yes | – | Yes | Gelastic Dacrystic |
| Study due to microcephaly and hypotonia | ||||||
| 4 | – | – | Yes | 5 | Yes Encephalopathy | Gelastic Dacrystic |
| 5 | Ultrasound 3rd trimester of gestation | – | Yes | – | Yes | Gelastic |
| Intracranial mass | ||||||
| 6 | – | – | Yes | – | – | – |
| 7 | – | – | Yes | – | – | – |
| 8 | – | – | Yes | 2.5 | Yes | No seizures |
| 9 | – | – | Yes | 5 | Yes | Gelastic |
| 10 | – | – | Yes | – | Yes | Gelastic |
TFUS, transfontanellar ultrasound; MRI, magnetic resonance imaging; CT, computed tomography; Video-EEG, video electroencephalogram.
The 8 patients who experienced seizures during the course of the disease underwent at least one conventional EEG and video-EEG monitoring study during sleep. All these patients showed abnormal results. The table lists the type of seizures recorded for each patient.
Treatment (Table 1)We first attempted to control seizures with pharmacological treatment in all patients with epilepsy. All patients required at least 2 different drugs for seizure control. The most frequently used drugs were oxcarbazepine (5), carbamazepine (4), topiramate (4), valproic acid (4), and levetiracetam (3). The patient with status gelasticus needed polytherapy with as many as 4 drugs.
Of the 10 patients, 7 underwent surgical removal of the hamartoma. In 6 patients, this was due to poor pharmacological control of the epileptic seizures, and in the remaining patient, due to the tumour increasing in size and exerting pressure on neighbouring structures. We ruled out surgery in 2 patients, since their seizures were controlled adequately by AEDs. The other patient was not referred for surgery as he did not experience seizures and the size of his tumour remained the same.
The initial technique used in 6 of the 7 patients who underwent surgery was resection. We achieved complete surgical resection in only one of these patients, while the 5 remaining patients underwent subtotal or partial resection. One of these last 5 patients needed 2 additional reoperations, which produced similar results. The seventh patient was selected to undergo gamma-knife surgery. We used this procedure as a second treatment approach for one of the cases with a subtotal surgical resection. As a result of a secondary hydrocephalus, 2 patients needed a ventriculoperitoneal shunt.
The mean time between onset of seizures and surgery was 5 years. Control over epileptic seizures was acceptable in 3 of the 8 patients; in one case, this had to do with the hamartoma resection.
DiscussionOur study is a retrospective review of 10 paediatric patients diagnosed with hypothalamic hamartoma in the last 20 years in a paediatric tertiary referral centre. The literature includes series with varying numbers of patients, but some of these series also include adult patients.17,27–29
The hypothalamus makes up less than 1% of the total brain volume, but it is a complex structure that includes numerous interconnections with both the cortex and the limbic neural networks. It regulates functions such as sleep, appetite, body temperature, reproduction, and sexual behaviour.30 It therefore plays an important role in modulating aggressive behaviours and in a wide variety of functions which are necessary for individual survival.
Hypothalamic hamartomas are non-neoplastic malformations of grey matter composed of hyperplastic neurons of different sizes.31
They are usually small lesions, measuring between 0.5 and 2cm in diameter and located at the base of the brain in the third ventricular floor, near the tuber cinereum and the mammillary bodies. These lesions may grow slowly in the interpeduncular cistern without displacing adjacent structures. It can take years before signs of compression appear.32 We did not find any single reason explaining why 2 patients’ hamartomas, measuring 4cm and 10cm respectively, were significantly larger than those described in the literature. However, a delay of approximately 4 years in diagnosing the lesion may explain the size of the largest hamartoma.
This disease's overall frequency is low, but the condition becomes serious if accompanied by epilepsy, behavioural and cognitive disorders, and/or precocious puberty (of which it is a rare cause). This is shown in the literature and in our paediatric series.1,2
There are two types of hypothalamic hamartomas, depending on their radiological imaging classification: pedunculated and sessile. Pedunculated or parahypothalamic hamartomas are connected to the third ventricular floor or suspended from the inferior hypothalamus by a peduncle. They are small or medium-sized, do not displace the hypothalamus, and are usually asymptomatic or provoke precocious puberty as the initial symptom. On the other hand, sessile or intrahypothalamic hamartomas surround and displace structures in the hypothalamus and the third ventricular wall. They are associated with epileptic seizures, cognitive disorders, psychomotor retardation, and psychiatric disorders.33–37 All patients in our series underwent a pituitary MRI scan to confirm the presence of a malformation. In 7 patients, this malformation was described as a sessile lesion causing a mass effect and/or displacing neighbouring structures (midbrain, third ventricle, optic tracts, optic chiasm, etc.).
MRI is much more sensitive than CT as a diagnostic test for this lesion. At times, it can even diagnose hamartoma when symptoms are not yet noticeable or even present, as was the case for patient 7, who was asymptomatic. The MRI scan should include examination of the hypothalamic and infundibular area and mammillary bodies.37,38
As mentioned above, epilepsy is one of the markers of this disease, and this is especially true for intrahypothalamic hamartomas. The mechanism of epileptogenesis is found in the microarchitecture of the hamartoma, which consists of small GABAergic neurons generating clusters which fire spontaneously.3–6,39,40 In our series, 80% of the patients had epilepsy, which was the first manifestation of hypothalamic hamartoma in all but one patient.
Gelastic seizures are the most common seizures in patients with hypothalamic hamartoma, especially in childhood, and almost always constitute the first epileptic manifestation.12,17,21 Nevertheless, they are often underdiagnosed, both clinically and using electrical activity testing, since seizures may go undetected or be mistaken for smiling, colic, or sleep disorders, especially in newborn and young unweaned babies. Moreover, many patients experience these seizures during sleep.41 This fact was confirmed in at least 2 of our patients who had been diagnosed initially with sleep disorders. All epileptic patients in our series experienced gelastic seizures at some point in the course of the disease. They were the first types of seizure to appear in more than half of the patients. In some descriptions of gelastic seizure cases, the seizures originated in the frontal lobe (cortical dysplasia). Therefore, where gelastic seizures are present, we should be aware that hypothalamic hamartoma is not the only possible aetiology.42–44 In our series, the mean age at onset of gelastic seizures was 13 months. In 1 patient, these seizures appeared in the first days of life, and in 2 others, at the age of 2 years; these data coincide with other studies in the literature in which researchers mention this type of seizure in the first days of life.8,9,45,46 In our series, 1 patient experienced status gelasticus (patient 4) and 3 patients experienced dacrystic seizures. These seizures have also been described in the literature.10,14,47–50
Many other types of seizures may appear in these patients, either at the beginning or during the course of the disease.9,12–14 In our series, we also found an additional type of seizure in 100% of the patients with seizures. Simple partial seizures were the most common, followed by complex partial seizures and generalised seizures (in some cases). Atonic seizures were only present in 1 patient. Progression of gelastic seizures to partial epilepsy usually occurs between the ages of 4 and 10. At times it may be difficult to differentiate between gelastic and partial seizures, as they have similar characteristics (decreased level of consciousness and orofacial automatisms) and both types of seizures may occur concomitantly.15,16,25,51 Generalised seizures also occur in patients with hypothalamic hamartoma, including tonic seizures, tonic–clonic seizures, and drop attacks.15–17,52 Some surgical series report a prevalence of generalised epilepsy of about 70%.15,53,54 Infantile spasms are rare in this disease, and did not appear in our series. However, they have been described in patients experiencing very early onset gelastic seizures during the neonatal period.21,55,56 In our series, the initial baseline EEG showed abnormal results in all patients. The most common abnormalities were unilateral or bilateral polyspike and polyspike-wave discharges with a focal onset and propagation to the frontotemporal regions. Such findings are common throughout the course of a patient's disease.15,17,18,21,22,52,57 Video EEG recorded gelastic seizure episodes in 7 patients and dacrystic seizures in 2 patients. EEG findings in one patient were described as typical of encephalopathy.
Only one patient had precocious puberty as the initial symptom, but as time passed, up to 50% of the patients developed that condition. This endocrine disorder has been described in several series of patients with hypothalamic hamartoma and gelastic seizures, with a frequency of 30%–40%.58–60 Its pathophysiology has not been fully clarified, but it is believed to involve an activation mechanism for the secretion of human luteinising hormone–releasing hormone (LH–RH).61 On the other hand, hamartoma is rarely associated with other endocrine diseases (growth deficit, diabetes insipidus, hypogonadism, etc.), unlike other hypothalamic diseases such as astrocytoma, glioma, and craniopharyngioma, which show a high incidence of endocrine disease. Nevertheless, our series contained one patient of short stature and another with panhypopituitarism.
Cognitive impairment (language delay, learning disability) and behavioural disorders (attention deficit hyperactivity disorder or ADHD, aggressiveness, anxiety, oppositional defiant disorder, etc.) are common in patients with epilepsy associated with hypothalamic hamartoma.1,35,62,63 Cognitive impairment/behavioural disorders have been linked to seizure severity and frequency. Nevertheless, this subject is controversial since some series show cognitive deficits to be present in patients before the onset of seizures.28 This is demonstrated by patients in our series, as they experienced associated comorbidities (psychomotor/mental retardation in 7 patients, learning disorders in 2, ADHD in 3 and aggressive/oppositional behaviours in 3). Multiple patients presented 2 or more of these problems.
As shown by several series of patients with epilepsy associated with hypothalamic hamartoma, gelastic seizures are resistant to AEDs and exert an effect on the cognitive and behavioural disorders present in these cases. This is true even when administering different drugs, high doses, and/or several drugs in polytherapy.20,56,60,64 We tried at least 2 AEDs with each patient in our series. Currently, only 2 patients have attained acceptable seizure control as a result of pharmacotherapy. One is taking oxcarbazepine monotherapy and the other, oxcarbazepine in combination with valproic acid.
For these reasons, patients with hypothalamic hamartoma who suffer from pharmacologically intractable epilepsy, progressive cognitive impairment, and/or behavioural problems are usually candidates for surgical treatment. This is their best option for eradicating seizures and improving cognitive and behavioural functions.24,45,64,65
In any case, we must be mindful of the potential risks of surgery (hypothalamic damage, memory loss, polyphagia or diabetes insipidus, and vascular damage55,57,28). Over the past few years, different surgical and non-surgical procedures have been used in order to minimise potential risks.23
Conventional surgical techniques, such as surgical resection and surgical excision through an inferior approach (craniotomy through transsylvian, subtemporal, or transfrontal approaches) or a superior approach (via transcallosal interfornical approach), deliver successful outcomes for seizure control, but surgical risks are high.34,56,64,66 Endoscopic disconnection is a safe alternative with good results.67,68 In our series, 6 patients underwent conventional surgery via different approaches. The tumour was completely resected in one case without any serious complications.
Nevertheless, emerging techniques, such as stereotaxic radiosurgery (especially gamma knife surgery), are being promoted as the first line of treatment of the near future. It has been shown that they produce successful outcomes with regard to seizure control and improvement of related disorders, and involve fewer risks than conventional surgery. At present, this technique is used as a second option when tumours persist after the initial conventional surgery.26,69,70 The only disadvantage of this procedure is the delay in obtaining therapeutic results; in most patients, seizures begin to subside approximately 6 months after surgery.44 In our series, 2 patients underwent surgery with this technique, one as an initial procedure and the other as a second attempt to remove the residual tumour after the initial surgery. We achieved acceptable seizure control in patient 2 only, the patient who underwent an apparently complete hamartoma resection. At present, patient 2 is on valproic acid monotherapy. However, progression of epilepsy in the other patients was not favourable; despite undergoing surgery, they required polytherapy in order to control their seizures. This contrasts with series of surgical patients in the literature which reported better results in the area of seizure control. This difference may be due to the fact that the hamartoma was not completely resected in most of our patients who underwent surgery.
In conclusion, we can state that hypothalamic hamartomas in our series have similar epidemiological and clinical characteristics to the hamartomas described in the literature. However, they present more difficulties for seizure control, whether by pharmacological or surgical means.
Conflicts of interestThe authors have no conflicts of interest to declare.
Please cite this article as: Castaño De La Mota C, et al. Hamartoma hipotalámico en la edad pediátrica: características clínicas, evolución y revisión de la literatura. Neurología. 2012;27:268–76.
This study was presented in the 35th Annual Meeting of the Spanish Society of Paediatric Neurology, Granada (2011), where it was chosen as one of the winning presentations.
