



Describir las características epidemiológicas, clínico-electroencefalográficas y la morbilidad asociada de los pacientes con hamartoma hipotalámico, así como la evolución y el tratamiento seguido.
Pacientes y métodosSe han revisado retrospectivamente las historias clínicas de 10 pacientes diagnosticados de hamartoma hipotalámico por resonancia magnética en los últimos 20 años.
ResultadosLa edad de debut de la epilepsia en los pacientes con hamartoma hipotalámico en nuestra serie está comprendida entre los primeros días de vida y los 2 años. De los 10 pacientes totales, 8 tuvieron crisis epilépticas en su evolución. Todos ellos presentaron crisis gelásticas, además de otros tipos de crisis, siendo las más frecuentes las parciales simples. Los hallazgos electroencefalográficos registrados fueron muy variables. Uno de los pacientes desarrolló encefalopatía epiléptica. Cinco pacientes presentaron algún tipo de trastorno de conducta. Cinco pacientes presentaron problemas cognitivos. En los 8 pacientes que presentaron crisis se ensayaron al menos 2 fármacos antiepilépticos diferentes y en 6 pacientes de estos se recurrió a alguna modalidad de tratamiento no farmacológica con el objetivo del control de las crisis. Solo en 3 de los 8 pacientes se ha conseguido aceptable control de su epilepsia. Cinco pacientes de la serie desarrollaron pubertad precoz. El tiempo medio de seguimiento de la serie es de 6 años.
ConclusionesLa epilepsia es la manifestación más frecuente de los hamartomas hipotalámicos, siendo en la mayoría de los casos farmacorresistente, lo que conlleva dificultades en el manejo de estos pacientes, precisando en muchas ocasiones cirugía para su control. Es frecuente la aparición de comorbilidad psiquiátrica y afectación cognitiva.
To describe the epidemiological and clinical-electroencephalographic characteristics, and associated morbidity of patients with hypothalamic hamartoma, as well as the treatment followed and outcomes
Patients and methodsWe have retrospectively reviewed the medical histories of 10 patients diagnosed with hypothalamic hamartoma by magnetic resonance imaging over the last 20 years.
ResultsThe age of onset of epilepsy in patients with hypothalamic hamartoma in our series was between the first days of life and 2 years. Of the 10 total patients, 8 had epileptic seizures during its progress. All of them had gelastic seizures, in addition to other types of seizures, with the most common being partial simple seizures. The electroencephalographic findings recorded were highly variable. One of the patients developed epileptic encephalopathy. Five patients had some kind of conduct disorder. Five patients had cognitive problems. At least 2 different antiepileptic drugs were measured in 8 of the patients who had seizures, and in 6 of these some type of non-pharmacological treatment had been used with the objective of seizure control. Only in 3 of 8 patients has been achieved Acceptable control of epilepsy had only been achieved in 3 out the 8 patients. Five patients of the series developed precocious puberty. The average time of follow-up of the series was approximately 6 years.
ConclusionsEpilepsy is the most frequent manifestation of hypothalamic hamartomas. Most cases were drug-resistant, which led to difficulties in the management of these patients, requiring surgery for their control on many occasions. Psychiatric comorbidity and cognitive impairment is common.
El hamartoma hipotalámico es una malformación no neoplásica que afecta al área hipotalámica localizada entre el tallo infundibular y los cuerpos mamilares, estando asociado a manifestaciones endocrinas y neurológicas. La prevalencia de este tumor en niños y adolescentes es aproximadamente de 1-2 casos/100.000 habitantes1. La mayoría de las ocasiones es esporádico, pero existe una rara asociación con el síndrome de Pallister-Hall, una enfermedad autosómica dominante que incluye malformaciones congénitas adicionales como polidactilia, ano imperforado y espina bífida o úvula bífida2.
Una de las principales características del hamartoma es su actividad epileptógena intrínseca, debido a la presencia de nódulos de pequeñas interneuronas gabaérgicas con actividad eléctrica espontánea3-6.
Las crisis gelásticas son las manifestaciones críticas más características y frecuentes en los pacientes con hamartoma hipotalámico, de inicio en los primeros años de vida (se han descrito incluso en recién nacidos), como episodios breves, estereotipados y frecuentes (en ocasiones en clusters) de risa inmotivada o automática, sin sensación de alegría, sin pérdida o con una breve disminución de la conciencia y generalmente acompañado de signos autonómicos (taquicardia, alteración de la respiración, rubicundez facial, dilatación pupilar, etc.)7-9. En algunos pacientes coexisten crisis gelásticas y dacrísticas o de llanto, en las cuales los pacientes pueden comenzar con un quejido, enrojecimiento facial que rápidamente desemboca en llanto y pudiendo asociarse automatismos orofaciales10. Se ha descrito en varias ocasiones en la literatura pacientes con estatus gelástico11-14.
En los pacientes con hamartoma hipotalámico se pueden observar otros tipos de manifestaciones epilépticas como crisis parciales complejas o crisis generalizadas9,12-14. Su desarrollo se atribuye a un proceso de epileptogénesis secundario15.
El electroencefalograma (EEG) de superficie tiene limitaciones para demostrar actividad epileptiforme en esta patología, debido a la localización profunda de esta lesión y a las complejas conexiones del hamartoma16. En las etapas iniciales de la enfermedad, el EEG intercrítico suele ser normal y las crisis gelásticas muestran una depresión difusa de la actividad de base17,18.
El espectro clínico del hamartoma hipotalámico es muy variable, desde pacientes con presencia asintomática del tumor o pacientes con alteraciones endocrinológicas aisladas como pubertad precoz, hasta pacientes con el síndrome descrito por Berkovic en 1988 de epilepsia gelástica de inicio precoz y hamartoma hipotalámico-pubertad precoz, caracterizado por encefalopatía epiléptica catastrófica, con problemas cognitivos y alteraciones severas del comportamiento19-22.
La epilepsia asociada al hamartoma hipotalámico es característicamente refractaria al tratamiento con fármacos antiepilépticos. Es excepcional conseguir un buen control de las crisis a pesar de la administración de dosis altas de antiepilépticos y utilizar asociaciones de estos1,11.
Se ha demostrado que la ablación del hamartoma ofrece al paciente la mejor opción para el control de las crisis cuando se ha comprobado que éstas son resistentes al tratamiento con fármacos antiepilépticos. Por otro lado, se consigue una mejoría en los problemas cognitivos y las alteraciones conductuales asociadas23,24.
Se han propuesto varias aproximaciones quirúrgicas para su exéresis (resección o desconexión por microcirugía, resección endoscópica); sin embargo, todas estas técnicas tienen un riesgo quirúrgico significativo. Por este motivo, en los últimos años se han desarrollado técnicas quirúrgicas no convencionales (radiocirugía con gamma-knife, implantación de semillas radiactivas, etc.) con aceptables resultados25,26.
Pacientes y métodosSe han revisado las historias clínicas de los pacientes registrados en nuestra base de datos con diagnóstico de hamartoma hipotalámico en los últimos 20 años (entre los años 1990 y 2010).
Se obtuvieron los datos epidemiológicos (edad, sexo, raza, embarazo, parto, periodo neonatal, antecedentes familiares), clínicos (edad al diagnóstico, modo de presentación, demora diagnóstica, comorbilidad asociada), exploraciones complementarias realizadas (EEG, video-EEG, resonancia magnética cerebral), valoración neuropsicológica y tratamientos recibidos.
En los casos que precisaron cirugía se recogieron los datos acerca del tipo de cirugía, edad de realización y complicaciones asociadas a esta.
Todos los pacientes incluidos estaban en edad pediátrica (0-12 años) en el momento del diagnóstico, y fueron seguidos en nuestro servicio por lo menos durante 6 años.
Todos los pacientes han completado el seguimiento tanto en las consultas de neuropediatría como neurocirugía y endocrinología cuando lo han precisado, no habiéndose perdido ningún paciente.
A los 5 pacientes a los que se les realizó valoración neuropsicológica, se les aplicaron tests cognitivos y del lenguaje adaptados a la edad.
ResultadosDatos epidemiológicos y perinatales (tabla 1)Seis de los 10 pacientes de nuestra serie son varones. Solo en un paciente hay antecedentes familiares de epilepsia. En cuanto a los antecedentes personales, el embarazo fue controlado en todos los pacientes excepto en uno. En otro paciente, en la ecografía del tercer trimestre de gestación ya se detectó una masa intracraneal por delante de cerebelo y por debajo del tálamo y tercer ventrículo. Al nacimiento precisaron ingreso 2 pacientes, uno por episodios paroxísticos (el paciente con diagnóstico prenatal por ecografía antes referido) y el otro por precisar cirugía de colocación de válvula de derivación ventrículo-peritoneal por hidrocefalia secundaria a quiste aracnoideo el sexto día de vida.
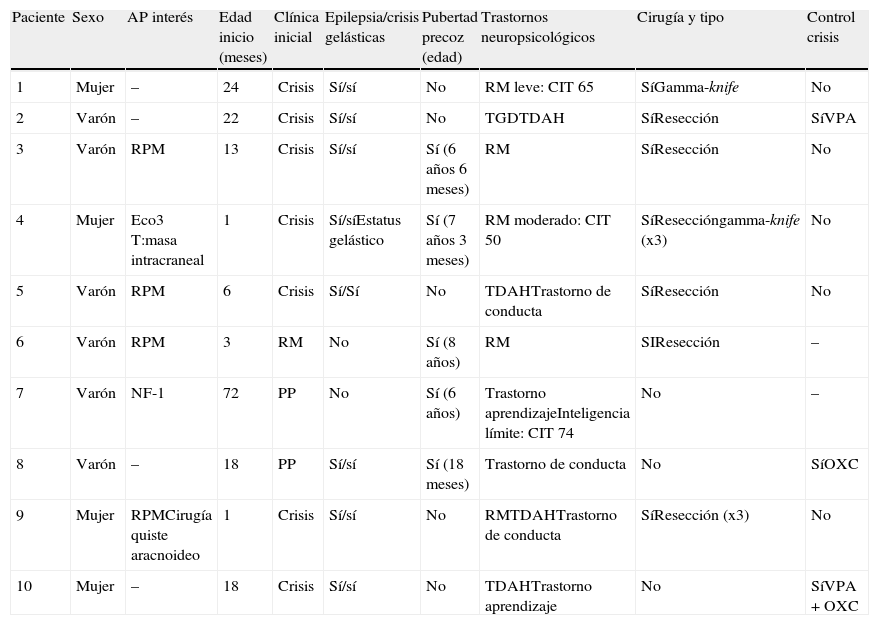
Resumen de las características de los pacientes con hamartoma hipotalámico de la serie
| Paciente | Sexo | AP interés | Edad inicio (meses) | Clínica inicial | Epilepsia/crisis gelásticas | Pubertad precoz (edad) | Trastornos neuropsicológicos | Cirugía y tipo | Control crisis |
| 1 | Mujer | – | 24 | Crisis | Sí/sí | No | RM leve: CIT 65 | SíGamma-knife | No |
| 2 | Varón | – | 22 | Crisis | Sí/sí | No | TGDTDAH | SíResección | SíVPA |
| 3 | Varón | RPM | 13 | Crisis | Sí/sí | Sí (6 años 6 meses) | RM | SíResección | No |
| 4 | Mujer | Eco3T:masa intracraneal | 1 | Crisis | Sí/síEstatus gelástico | Sí (7 años 3 meses) | RM moderado: CIT 50 | SíReseccióngamma-knife (x3) | No |
| 5 | Varón | RPM | 6 | Crisis | Sí/Sí | No | TDAHTrastorno de conducta | SíResección | No |
| 6 | Varón | RPM | 3 | RM | No | Sí (8 años) | RM | SIResección | – |
| 7 | Varón | NF-1 | 72 | PP | No | Sí (6 años) | Trastorno aprendizajeInteligencia límite: CIT 74 | No | – |
| 8 | Varón | – | 18 | PP | Sí/sí | Sí (18 meses) | Trastorno de conducta | No | SíOXC |
| 9 | Mujer | RPMCirugía quiste aracnoideo | 1 | Crisis | Sí/sí | No | RMTDAHTrastorno de conducta | SíResección (x3) | No |
| 10 | Mujer | – | 18 | Crisis | Sí/sí | No | TDAHTrastorno aprendizaje | No | SíVPA+OXC |
AP: antecedentes personales; OXC: oxcarbacepina; PP: pubertad precoz; RM: retraso mental; RPM: retraso psicomotor; TDAH: trastorno por déficit de atención e hiperactividad; TGD: trastorno generalizado del desarrollo; VPA: ácido valproico.
En 4 pacientes existe una historia de retraso del desarrollo psicomotor desde las primeras fases. Una paciente tenía un diagnóstico previo de neurofibromatosis tipo 1.
Forma de presentación (tabla 1)La edad de inicio de los síntomas fue muy variable: desde los primeros días de vida en 2 pacientes de la serien hasta los 6 años en el paciente en el cual el hamartoma fue un hallazgo casual tras realizar una resonancia magnética (RM) para valoración de neurofibromatosis tipo 1.
La edad de derivación de estos pacientes para inicio de estudio o seguimiento en consulta de neurología también fue variable (entre los 2 y los 14 años) y el tiempo de seguimiento medio de la serie es de 6 años. Tres pacientes fueron dados de alta por haber cumplido la mayoría de edad.
Las manifestaciones clínicas iniciales de los pacientes con hamartoma hipotalámico fueron: en 7 crisis epilépticas, pubertad precoz en 2 y retraso psicomotor en otro. Dos pacientes de la serie no presentaron crisis en toda su evolución. La edad de inicio de las crisis osciló entre los primeros días de vida y los 4 años de edad, con una media de 10 meses.
Las crisis gelásticas fueron comunes en todos los pacientes con epilepsia, siendo el primer tipo de crisis en aparecer en 5 de los mismos; en 2 pacientes estas aparecieron en los primeros días de vida. Un paciente presentó estatus gelástico a los 11 años. Dos pacientes presentaron además crisis dacrísticas, siendo en uno de ellos el episodio crítico inicial. Todos los pacientes de la serie desarrollaron además otro tipo de crisis, siendo las más frecuentes parciales simples, seguidas de parciales complejas y generalizadas. En un paciente se constataron crisis atónicas.
Evolución (tabla 1)Cinco pacientes desarrollaron pubertad precoz en algún momento de su evolución, siendo todos ellos varones excepto una. Dos de estos pacientes tenían además déficit de hormona de crecimiento y, a su vez, uno de ellos hipotiroidismo.
Seis pacientes de la serie presentaron algún tipo de retraso madurativo o problemas de aprendizaje y un paciente tuvo una evolución a trastorno generalizado del desarrollo. Se realizó una evaluación neuropsicológica a 5 pacientes, obteniéndose un cociente intelectual (CIT) dentro de límites normales en 2 de ellos, inteligencia límite en 1, retraso mental leve en 1 y retraso mental moderado en el último.
Cinco pacientes presentaron trastornos de conducta: 4 casos déficit de atención y/o hiperactividad y 2 sujetos conductas agresivas, coexistiendo en uno de ellos ambas patologías.
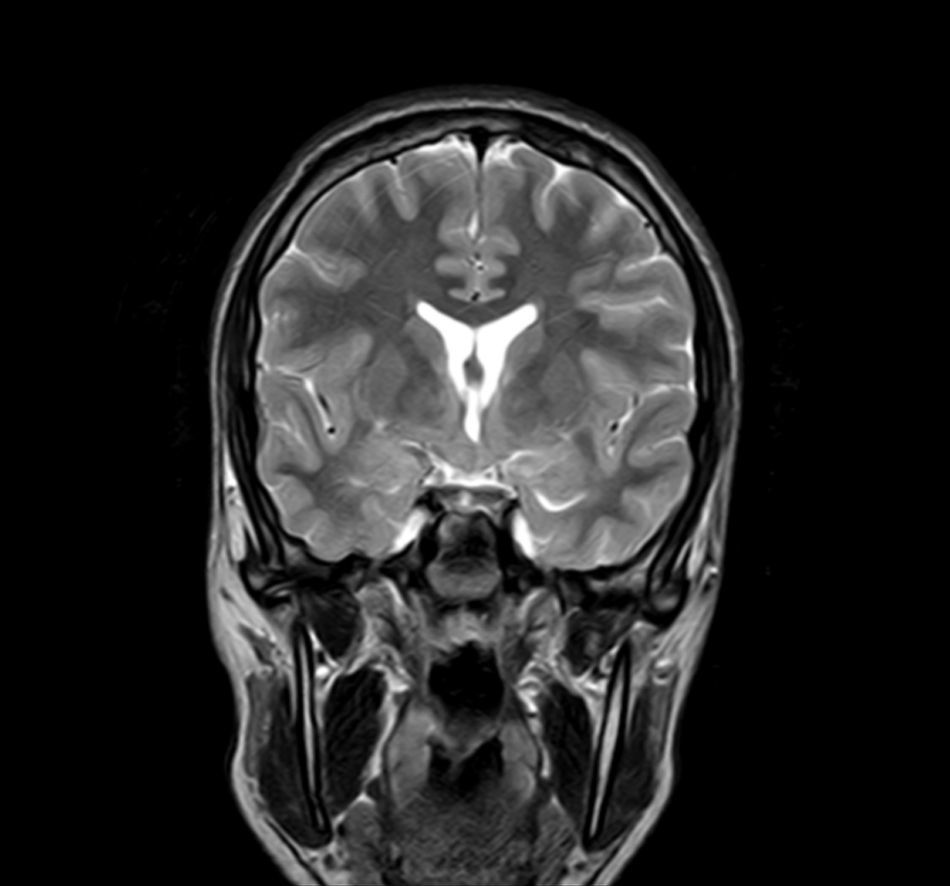
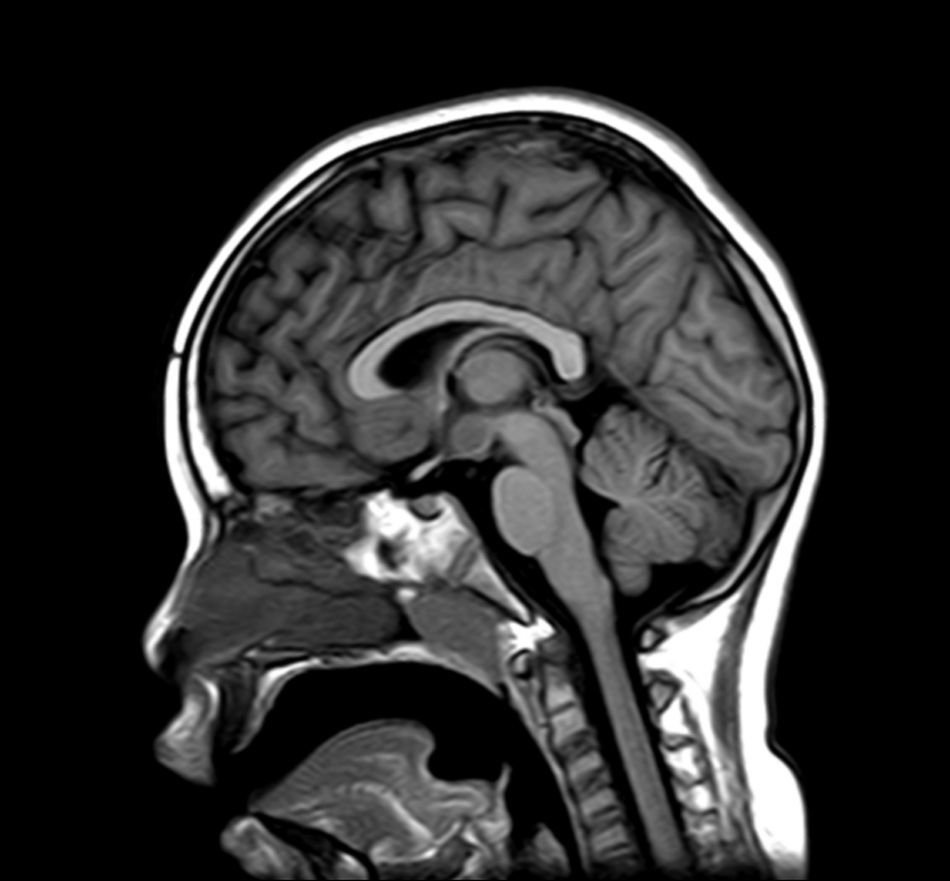
Exploraciones complementarias (tabla 2)En cuanto a las pruebas de imagen realizadas, en 3 pacientes se realizó ecografía transfontanelar por diferentes motivos. Sólo se realizo tomografía computarizada (TC) en un paciente. La RM con cortes de hipófisis fue la prueba diagnóstica que confirmó la presencia del hamartoma hipotalámico en todos los pacientes de la serie (figs. 1 y 2), oscilando el tamaño en RM entre 1,3cm y 10cm.


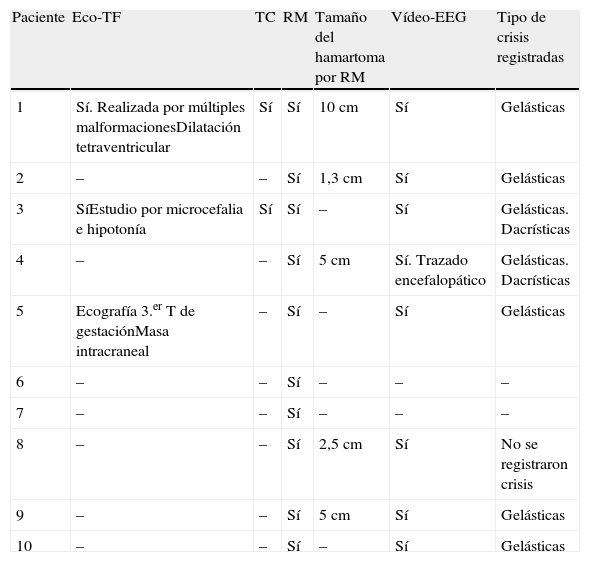
Resumen de las pruebas complementarias realizadas y hallazgos de estas
| Paciente | Eco-TF | TC | RM | Tamaño del hamartoma por RM | Vídeo-EEG | Tipo de crisis registradas |
| 1 | Sí. Realizada por múltiples malformacionesDilatación tetraventricular | Sí | Sí | 10cm | Sí | Gelásticas |
| 2 | – | – | Sí | 1,3cm | Sí | Gelásticas |
| 3 | SíEstudio por microcefalia e hipotonía | Sí | Sí | – | Sí | Gelásticas. Dacrísticas |
| 4 | – | – | Sí | 5cm | Sí. Trazado encefalopático | Gelásticas. Dacrísticas |
| 5 | Ecografía 3.er T de gestaciónMasa intracraneal | – | Sí | – | Sí | Gelásticas |
| 6 | – | – | Sí | – | – | – |
| 7 | – | – | Sí | – | – | – |
| 8 | – | – | Sí | 2,5cm | Sí | No se registraron crisis |
| 9 | – | – | Sí | 5cm | Sí | Gelásticas |
| 10 | – | – | Sí | – | Sí | Gelásticas |
Eco-TF: ecografía transfontanelar; RM: resonancia magnética; TC: tomografía computarizada; vídeo-EEG: videoelectroencefalograma.
Los 8 pacientes que presentaron crisis en su evolución tienen al menos un estudio de EEG convencional y otro de monitorización con vídeo-EEG de sueño. Presentaron alteraciones en el registro todos los pacientes. En la tabla constan el tipo de crisis registradas en cada paciente.
Tratamiento (tabla 1)En todos los pacientes con epilepsia se intentó inicialmente control farmacológico de las crisis, necesitándose en todos ellos al menos 2 fármacos diferentes con el objetivo del control de las crisis. Los fármacos más utilizados fueron: oxcarbacepina (5), carbamazepina (4), topiramato (4), ácido valproico (4) y levetiracetam (3). En el paciente que presentó el estatus gelástico se asociaron hasta 4 fármacos.
En 7 de los 10 pacientes se llevó a cabo la cirugía del hamartoma: en 6 de ellos por mal control de las crisis con antiepilépticos y en el último por aumento del tamaño del tumor con compresión secundaria de las estructuras vecinas. En 2 pacientes no se ha planteado la cirugía, ya que hasta el último seguimiento el control de las crisis con antiepilépticos es aceptable. En el otro paciente no se planteó cirugía por la ausencia de crisis y estancamiento del tamaño del tumor.
La técnica quirúrgica inicial fue la cirugía de resección en 6 pacientes de los 7 pacientes intervenidos, lográndose resección completa solo en uno de ellos, mientras que en los 5 restantes la resección conseguida fue subtotal o parcial. Uno de estos últimos pacientes se reintervino posteriormente en dos ocasiones más, con resultados similares. En el séptimo paciente, el gamma-knife fue la otra modalidad quirúrgica elegida. Esta técnica se utilizó como segunda aproximación terapéutica en uno de los pacientes en los cuales la resección fue subtotal. Dos pacientes precisaron la colocación de una válvula de derivación ventrículo-peritoneal por hidrocefalia secundaria.
El tiempo medio transcurrido entre el inicio de las crisis y la cirugía fue de 5 años. Se ha logrado un aceptable control de las crisis epilépticas solo en 3 de los 8 pacientes, en uno de ellos en relación con la resección del hamartoma.
DiscusiónEn este trabajo se presenta una revisión retrospectiva de 10 pacientes pediátricos diagnosticados de hamartoma hipotalámico en los últimos 20 años en un hospital infantil de tercer nivel. Hay varias series descritas en la literatura con un número de pacientes variable, pero muchas de ellas incluyen también a pacientes adultos17,27-29.
El tamaño del hipotálamo es menos del 1% del volumen total cerebral, pero es una estructura compleja que tiene interconexiones muy numerosas tanto con la corteza como con los circuitos neuronales límbicos, comportándose como una estructura integradora de funciones como el sueño, el apetito, la termorregulación, la reproducción y la conducta sexual30. Por tanto, parece desempeñar un papel importante en la modulación de las conductas agresivas y en gran variedad de funciones necesarias para la supervivencia del individuo.
Los hamartomas hipotalámicos son malformaciones no neoplásicas de sustancia gris compuestas de neuronas hiperplásicas de distinto tamaño31.
Habitualmente, se trata de una lesión pequeña, entre 0,5 y 2cm de diámetro, localizada en la base del cerebro en el piso del tercer ventrículo, cerca del tuber cinereum y los cuerpos mamilares. Un crecimiento lento dentro del espacio interpeduncular puede ocurrir sin desplazar las estructuras adyacentes y pueden transcurrir años antes de que existan signos de compresión32. No hemos podido encontrar una causa común que justifique el hecho de que el tamaño de los hamartomas de dos de los pacientes de nuestra serie (4cm y 10cm, respectivamente) sean significativamente más grandes que los referidos en la literatura; sin embargo, sí que existe una demora diagnóstica de aproximadamente 4 años que podría justificar el tamaño de la lesión en el paciente con el hamartoma más grande.
Su frecuencia global es baja, pero su importancia radica en su asociación con epilepsia, problemas cognitivos y conductuales y/o pubertad precoz (siendo una causa rara de la misma) como se demuestra en la literatura y en nuestra serie pediátrica1,2.
Existen dos tipos de hamartomas hipotalámicos, según su clasificación radiológica: pedunculados y sésiles. Los pedunculados o parahipotalámicos están anexos al suelo del tercer ventrículo o suspendidos desde el hipotálamo inferior por un pedúnculo, no desplazan el hipotálamo, son de pequeño o mediano tamaño y suelen ser asintomáticos o debutar como pubertad precoz. Por otra parte, los hamartomas sésiles o intrahipotalámicos engloban y desplazan estructuras del hipotálamo y la pared del tercer ventrículo, asociando crisis epilépticas, alteraciones cognitivas, retraso psicomotor y problemas psiquiátricos33-37. A todos los pacientes de nuestra serie se les realizó una RM craneal con cortes de hipófisis, en la que se confirmó la existencia de la malformación. En 7 pacientes aparece descrito como una lesión de tipo sésil, produciendo efecto masa y/o desplazando estructuras vecinas (mesencéfalo, tercer ventrículo, cintillas ópticas, quiasma óptico, etc.).
La RM es una prueba de imagen mucho más sensible que la TC para la detección de esta lesión, pudiendo en ocasiones diagnosticar el hamartoma en ausencia de clínica asociada o antes de que esta aparezca, como ocurrió en nuestro paciente 7 asintomático en relación con esta lesión. La RM debe incluir exploración del área hipotalámica, infundibular y de los cuerpos mamilares37,38.
La epilepsia es uno de los marcadores de esta enfermedad, como nos hemos referido anteriormente, más frecuentemente en relación con el tipo intrahipotalámico. El mecanismo de epileptogénesis subyace en la microarquitectura del hamartoma, compuesto por pequeñas neuronas gabaérgicas que forman nódulos y que tienen actividad eléctrica espontánea3-6,39,40. El 80% de los pacientes de nuestra serie presentaron epilepsia, siendo la primera manifestación del hamartoma hipotalámico en todos ellos, excepto en uno.
Las crisis gelásticas son las más frecuentes en el hamartoma hipotalámico, sobre todo en la infancia, y son casi siempre la primera manifestación epiléptica12,17,21. Sin embargo, muchas veces son infradiagnosticadas, tanto clínica como eléctricamente, al pasar desapercibidas o confundirse con sonrisa, cólicos del lactante o trastornos del sueño, de forma más frecuente en los recién nacidos o lactantes pequeños. Además, en un porcentaje alto tienen lugar durante el sueño41. Este hecho se constató en al menos 2 de nuestros pacientes, clasificados como trastornos del sueño en un primer momento. Todos los pacientes con epilepsia de nuestra serie presentaron crisis gelásticas en algún momento de su evolución, y en más de la mitad de los pacientes, fueron las primeras en aparecer. Se han descrito casos de crisis gelásticas en las cuales su origen estaba en el lóbulo frontal (displasia cortical). Por tanto, ante la presencia de crisis gelásticas no hay que descartar otras posibles etiologías distintas al hamartoma hipotalámico42-44. La edad media de inicio de las crisis gelásticas en nuestra serie fue de 13 meses, existiendo un paciente en el cual aparecieron en los primeros días de vida, y en 2 de ellos a los 2 años; estos datos coinciden con otras series de la literatura, habiéndose descrito este tipo de crisis en el primer día de vida8,9,45,46. En nuestra serie hubo un paciente que presentó un estatus gelástico (paciente 4) y 3 pacientes con crisis dacrísticas, entidades ya descritas en la literatura10,14,47-50.
Otros muchos tipos de crisis pueden aparecer en estos pacientes, bien al inicio o en la evolución de la enfermedad9,12-14. En nuestra serie de pacientes, en el 100% de los que presentaron crisis se objetivo algún tipo de crisis distintas: parciales simples las más frecuentes, seguidas de parciales complejas y un algún caso generalizadas, constatándose crisis atónicas solo en uno de ellos. La evolución de las crisis gelásticas a una epilepsia parcial (con o sin generalización secundaria) suele tener lugar entre los 4 y 10 años de edad; la diferenciación entre crisis gelásticas y parciales complejas puede ser difícil en ocasiones, por tener elementos similares (disminución del nivel de consciencia, automatismos orofaciales) y por poder ocurrir de forma concomitante ambos tipos de crisis15,16,25,51. También se han descrito crisis generalizadas en los pacientes con hamartoma hipotalámico, incluyendo crisis tónicas, tónico-clónicas y drop attacks15-17,52. En algunas series quirúrgicas la prevalencia de epilepsia generalizada se sitúa en un 70%15,53,54. Los espasmos infantiles son poco frecuentes en esta patología, y ausentes en nuestra serie, pero sí se han descrito en pacientes en los cuales las crisis gelásticas aparecieron de forma muy precoz en el período neonatal21,55,56. En nuestra serie, el EEG basal inicial presentaba alteraciones en todos los pacientes. Las más frecuentes fueron polipuntas y polipunta-onda de inicio focal y con propagación a regiones fronto-temporales, uni o bilaterales, hallazgos frecuentes en la evolución de estos pacientes15,17,18,21,22,52,57. En 7 pacientes se registraron clínicamente durante el vídeo-EEG algún episodio de crisis gelásticas y en 2 pacientes crisis dacrísticas. En uno de los pacientes describen el registro como encefalopático.
La pubertad precoz fue la clínica inicial solo en uno de nuestros pacientes, pero evolutivamente hasta el 50% la desarrollaron. Este trastorno endocrino es un hallazgo que se describe en varias series pacientes con hamartoma hipotalámico y crisis gelásticas con una frecuencia del 30-40%58-60. La fisiopatología no está plenamente aclarada, pero se postula un mecanismo activador en la secreción de la hormona luteinizante humana LH-RH61. Por otro lado, el hamartoma no se suele asociar a otras alteraciones endocrinas (déficit de crecimiento, diabetes insípida, hipogonadismo, etc.), en contraste con otras patologías hipotalámicas como astrocitomas, gliomas o craneofaringiomas, en los cuales su incidencia es elevada. Sin embargo, en nuestra serie existe un paciente con talla baja y otro con panhipopituitarismo.
El deterioro cognitivo (retraso del lenguaje, dificultades de aprendizaje) y los trastornos de conducta (trastorno por déficit de atención e hiperactividad o TDAH, agresividad, ansiedad, trastorno desafiante, etc.) son frecuentes en los pacientes con epilepsia asociada a un hamartoma hipotalámico1,35,62,63. Se han correlacionado con la frecuencia y la severidad de las crisis, pero existe debate en este punto ya que en algunas series de pacientes se ha demostrado que los déficits cognitivos ya existen antes del inicio de las crisis64. Esto se refleja en los pacientes de nuestra serie, ya que todos ellos presentaron comorbilidad asociada (retraso psicomotor/mental en 7, trastornos de aprendizaje en 2, TDAH en 3 y conductas agresivas/oposicionistas en 3), coincidiendo 2 o más de estos problemas en más de un paciente.
Como se ha podido demostrar en varias series de pacientes con epilepsia asociada a hamartoma hipotalámico, las crisis gelásticas son refractarias al tratamiento con fármacos antiepilépticos (FAE), además de influir sobre el deterioro cognitivo y conductual que ocurre en ellos. Esto ocurre a pesar de la utilización de diferentes fármacos, altas dosis y/o con asociación de varios de los mismos20,56,60,65. En los pacientes de nuestra serie se ensayaron al menos 2 FAE en cada uno de ellos, actualmente con aceptable control de crisis con farmacoterapia solo en 2 pacientes, uno de ellos en monoterapia con oxcarbacepina y otro con la combinación de esta con valproato.
Por estos motivos, los pacientes con hamartoma hipotalámico que presentan epilepsia intratable farmacológicamente, deterioro cognitivo progresivo y/o problemas conductuales, normalmente son candidatos para un tratamiento quirúrgico. Este les ofrece la mejor oportunidad para erradicar las crisis y mejorar la función cognitiva y conductual24,45,65,66.
No hay que obviar los posibles riesgos quirúrgicos (daño hipotalámico, problemas de memoria, hiperfagia o diabetes insípida, y daño vascular55,57,64). Con el objetivo de minimizar estos posibles riesgos, se han utilizado diferentes procedimientos quirúrgicos y no quirúrgicos en los últimos años23.
Las técnicas quirúrgicas convencionales, como son la resección o exéresis quirúrgica por abordaje inferior (realizando craneotomía transilviana, subtemporal, transfrontal) o superior (aproximación transcalloso-interfornical), tienen buenos resultados de control de crisis, pero con riesgos quirúrgicos elevados34,56,65,67. Una alternativa segura es la desconexión quirúrgica endoscópica, con buenos resultados68,69. Seis de los pacientes de la serie se sometieron a cirugía convencional mediante abordajes diferentes, con resección tumoral total solo en uno de ellos, sin describirse complicaciones graves asociadas a esta.
Sin embargo, son las nuevas técnicas emergentes como la radiocirugía estereotáctica (concretamente la cirugía con gamma-knife), las que se postulan como tratamiento de primera línea en un futuro próximo, ya que han demostrado buenos resultados de control de crisis y mejoría de los trastornos asociados, así como una disminución en los riesgos que supone la cirugía convencional. Actualmente también se está usando esta técnica como segunda opción ante persistencia de restos tumorales tras una primera cirugía convencional26,70,71. La única desventaja de este procedimiento es el retraso en el efecto, ya que en la mayoría de los pacientes las crisis comienzan a controlarse alrededor de 6 meses después del procedimiento44. Dos pacientes de la serie fueron intervenidos con esta técnica, en uno de ellos como procedimiento inicial y en el otro como una segunda aproximación en el intento de extirpar los restos tumorales tras una primera cirugía. Solo en el paciente 2 en el que se realizó resección aparentemente completa del hamartoma se ha conseguido un aceptable control de las crisis, en la actualidad en monoterapia con ácido valproico. En el resto de los pacientes, sin embargo, la evolución de la epilepsia no fue favorable, precisando politerapia para intentar el control de las crisis a pesar de la cirugía realizada. Esto contrasta con las series de pacientes quirúrgicos referidos en la literatura, con mejores resultados en cuanto a control de las crisis. Esta diferencia puede explicarse porque en la mayoría de los pacientes de nuestra serie que se sometieron a cirugía, no se logró una resección completa del hamartoma.
Como conclusión, podemos afirmar que los hamartomas hipotalámicos de nuestra serie se comportan de manera similar a los descritos en la literatura en cuanto a características epidemiológicas y clínicas, pero con mayores dificultades para un control de las crisis, tanto farmacológicas como quirúrgicas.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.
Trabajo presentado en la última Reunión Anual de la Sociedad Española de Neurología Pediátrica, Granada. Elegida como una de las comunicaciones-premio en la XXXV Reunión Anual Granada 2011.

