



El síndrome de desmielinización osmótica (SDO) es una enfermedad neurológica grave e infrecuente que incluye la mielinólisis central pontina (MCP) y extrapontina, siendo la forma pontina la más habitual. Fue descrito por primera vez por Adams et al. en 19591. Tradicionalmente ha sido observado en pacientes alcohólicos, con trasplante hepático y en estados hiperosmolares (en particular secundario a la corrección rápida de hiponatremia crónica). Sin embargo, se han descrito en situaciones de estado hiperosmolar grave sin hiponatremia asociada, incluyendo diabetes mellitus, hipofosfatemia grave, hipopotasemia, insuficiencia renal, hemodiálisis, hiperémesis gravídica, anorexia nerviosa, enfermedad de Wilson, quemaduras graves, lupus eritematoso sistémico, porfiria intermitente aguda, hepatitis por citomegalovirus, síndrome hemofagocítico asociado al de virus Epstein-Barr, shock anafiláctico y golpe de calor entre otros2,3. En general, parece que cualquier alteración brusca del estado hiperosmolar puede precipitar esta entidad.
Se presenta el caso de un paciente varón de 52 años con antecedentes personales de diabetes mellitus tipo 2 con complicaciones microvasculares asociadas, mal control metabólico (último registro de Hb glucosilada 11,7% hace 4 meses), dislipemia, exconsumidor de drogas por vía parenteral (sin otros hábitos tóxicos), infección por VIH categoría A-3 con buena situación inmunovirológica y cirrosis hepática descompensada por VHC genotipo-3 curada (con sofosbuvir y daclatasvir), pero con persistencia de complicaciones graves en los últimos meses (episodios de encefalopatía hepática, hemorragia digestiva varicosa y descompensación hidrópica) a consecuencia de una cirrosis muy evolucionada, pendiente de valoración para inclusión en lista de trasplante hepático.
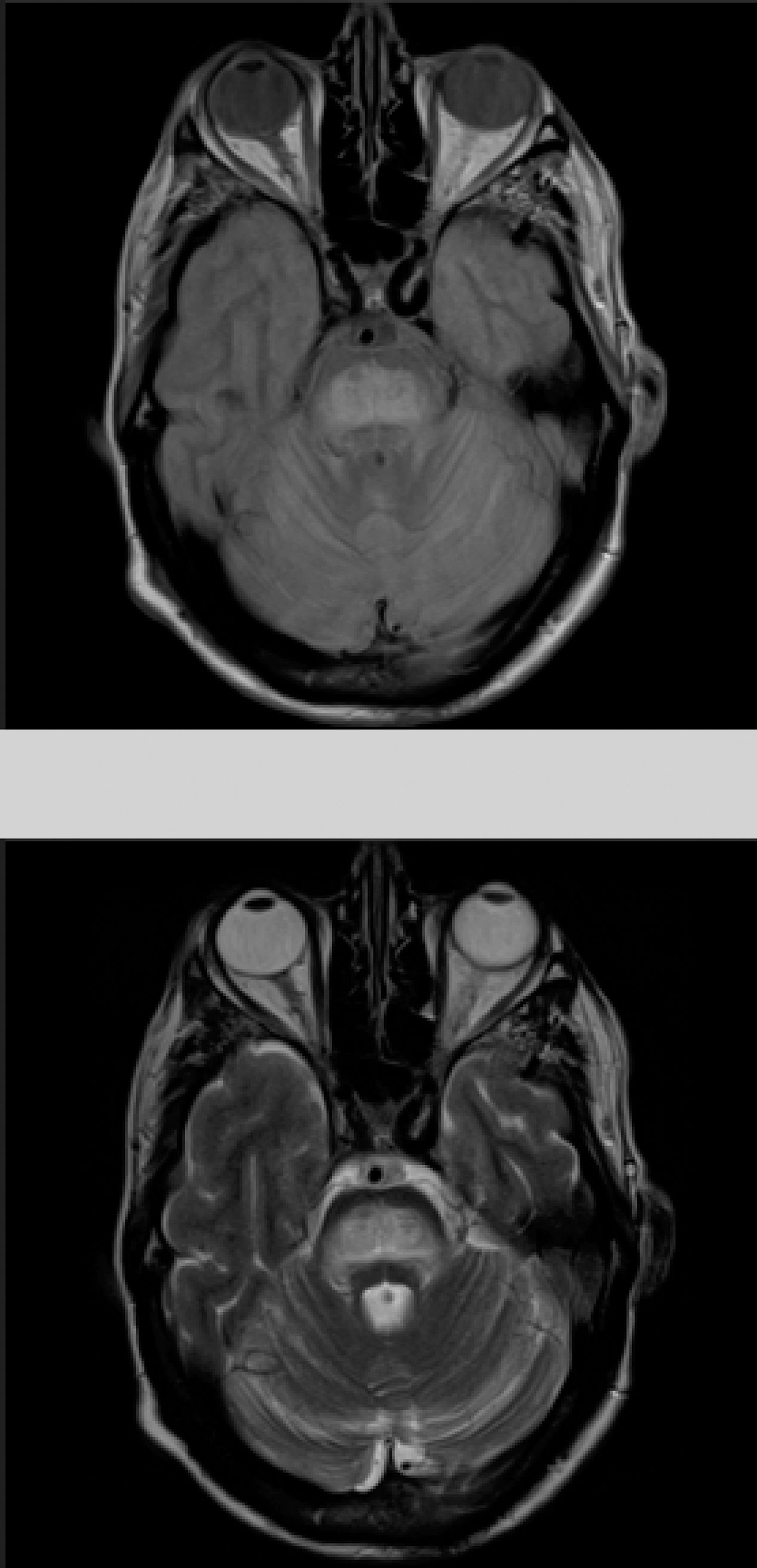
El paciente comienza con un cuadro de 2 meses de evolución consistente en disartria, disfagia e inestabilidad para la marcha. En la exploración se objetiva disartria moderada, limitación para la supraversión de la mirada y ataxia troncular intensa. Los exámenes de laboratorio a su ingreso mostraban, glucosa 406mg/dl; creatinina 1,37; Na 133mmol/l; K 4,62mmol/l, siendo el resto de resultados irrelevantes. Se realizó una tomografía computarizada cerebral sin hallazgos significativos y análisis del líquido cefalorraquídeo en las primeras 24h del ingreso con estudio bioquímico (incluidas bandas oligoclonales), citológico y microbiológico (PCR virus herpes 1 y 2, antígenos para listeria, meningococo, neumococo, Haemophilus, enterovirus, Epstein-Barr, virus varicela zóster, citomegalovirus, hongos, carga viral VIH y micobacterias) todos ellos sin significación patológica. La resonancia magnética (RM) cerebral (fig. 1) realizada a las 48h del ingreso mostraba una lesión hiperintensa en las secuencias DP-T2 y FLAIR en la protuberancia, con discreta restricción en estudio de difusión, todo ello compatible con MCP.
 en protuberancia, que presenta discreta restricción en el estudio de difusión que en el contexto clínico del paciente sugestivo de mielinólisis central pontina.")
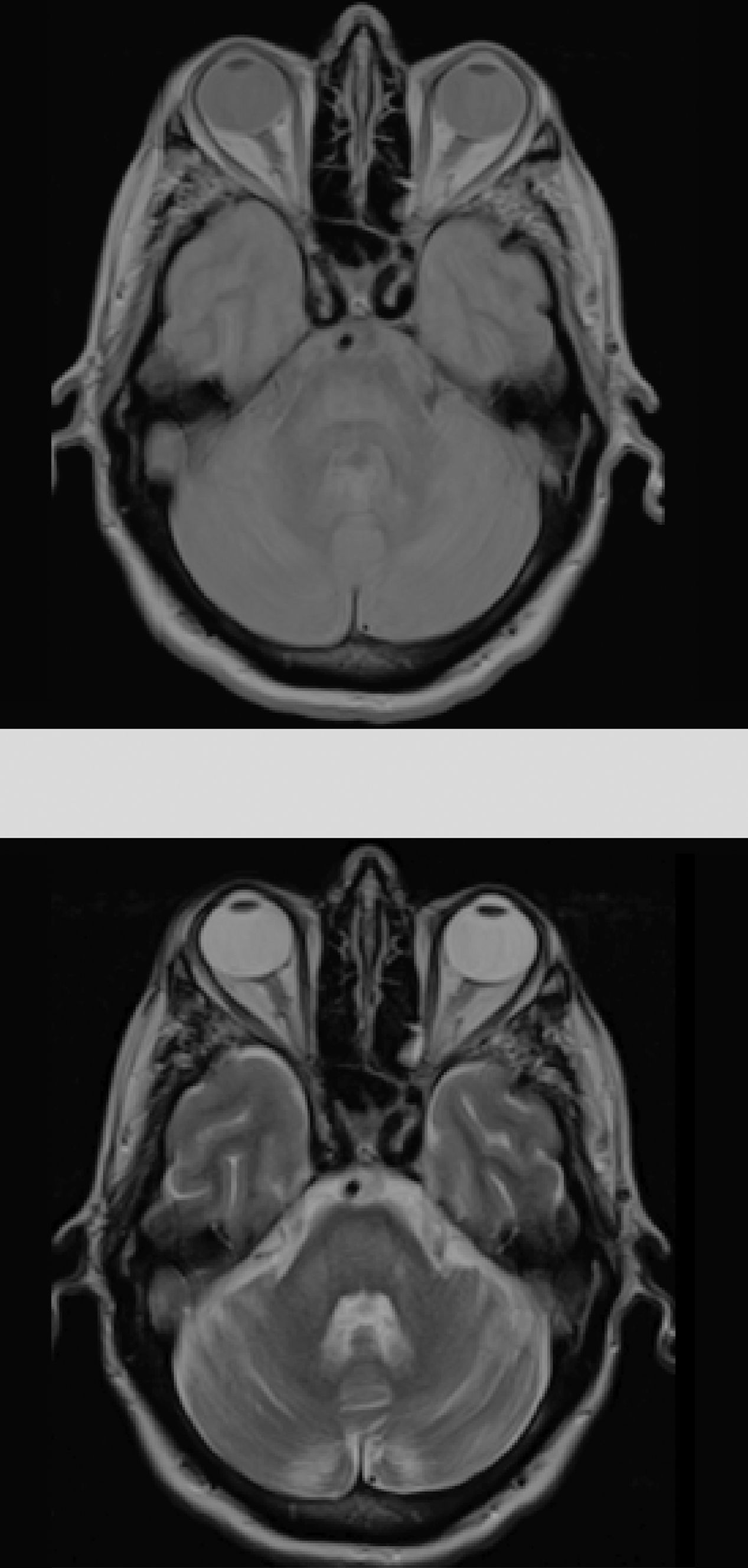
Dados los antecedentes del paciente se consideró haber presentado una hiponatremia corregida que no fue confirmada en los análisis previos, destacando únicamente persistentes cifras elevadas de glucemias (en torno 500-600mg/dl), lo que condicionaría un estado hiperosmolar como desencadenante de la MCP. El resto de parámetros analíticos fueron normales (bioquímica, iones, autoinmunidad, marcadores tumorales, antineuronales, tóxicos). Durante su ingreso se consiguió buen control de las glucemias con progresiva mejoría neurológica encontrándose al alta solo con disartria leve. Fue reevaluado 5 meses después objetivando resolución completa de la clínica neurológica y mostrando en la RM cerebral evidente mejoría de las lesiones (fig. 2).
 protuberancial como secuela de mielinólisis central pontina.")
Los mecanismos fisiopatológicos no están bien establecidos en la actualidad, pero se postula que son producidos como consecuencia de la rotura de la barrera hematoencefálica (BHE) secundario a situaciones de estrés osmótico, dando lugar a la desmielinización y apoptosis de los oligodendrocitos4.
Ashrafian y Davey5 han argumentado que la etiología es multifactorial siendo más probable que ocurra en pacientes con condiciones que predisponen a deficiencias en el suministro de energía en las neuronas y células gliales. Observando que los pacientes con corrección lenta de la hiponatremia pueden desarrollar MCP si los problemas electrolíticos ocurren en un marco de estados de privación de energía como el alcoholismo crónico o la enfermedad hepática. Estos autores razonaron que los alcohólicos o los pacientes cirróticos, así como pacientes con hipoglucemia de otras causas, pueden carecer de una reserva suficiente de glucosa o glucógeno para suministrar a las células gliales, necesaria para mantener la actividad de la bomba de Na-K-ATPasa, mecanismo responsable del transporte de electrones en el cerebro5. La deficiencia subclínica de tiamina puede también exacerbar el problema porque desciende la captación de glucosa en el cerebro y disminuye las fuentes de energía disponibles para las células gliales. Las neuronas pueden liberar glutamato y otras moléculas excitatorias en respuesta al estrés osmótico, aumentando el contenido de calcio intracelular. Este último estímulo sería el causante de la muerte celular apoptótica, que estos autores proponen pueda ser el mecanismo final para la MCP5.
Las manifestaciones clínicas son muy heterogéneas, desde formas asintomáticas hasta trastornos neurológicos severos como disfagia, disartria, tetraparesia, coma, trastornos neuropsiquiátricos como inquietud, labilidad emocional, apatía, mutismo, agitación, desinhibición y trastornos del movimiento como parkinsonismo o distonías entre otros3,6. Habitualmente la presentación inicial más frecuente la encefalopatía2. Las lesiones pontinas/extrapontinas pueden aparecer aisladas o superponerse en el tiempo, siendo las manifestaciones extrapontinas (por sí solas o combinadas con las pontinas) las menos frecuentes6. Se ha descrito recuperación completa incluso en aquellos pacientes con una presentación neurológica severa2,3.
El gold standard para el diagnóstico es la RM cerebral que ha permitido detectar incluso los casos asintomáticos y con ello mejorar el pronóstico de esta entidad2. Hay que tener en cuenta que los hallazgos radiológicos pueden no observarse dentro de la primera semana de la clínica, siendo necesario repetir la prueba de imagen si la sospecha es alta6.
Se ha ensayado en animales el tratamiento precoz con dexametasona (DXT) tras la corrección rápida de la hiponatremia con excelentes resultados clínicos y mejoría del pronóstico basándose en la capacidad de la DXT para regular y evitar el daño de la BHE y disminuir la liberación de citocinas por la microglía7. Sin embargo, por el momento no existen ensayos controlados en humanos.
Este caso es de especial interés por algunos aspectos peculiares. En primer lugar un curso de evolución subaguda, la hiperglucemia aislada como principal factor desencadenante y la resolución completa de la clínica tras conseguir un buen control metabólico en un paciente que mostraba como factor condicionante descrito la cirrosis2. La mayoría de los casos de MCP que se han publicado presentan situaciones de hiperglucemia con cetoacidosis concomitante, anomalía en la natremia o tras el tratamiento del estado hiperglucémico hiperosmolar8–10, y de manera menos frecuente secundario a hiperglucemia aislada8,11,12. Así pues, con este caso se apoyaría la hipótesis de que fluctuaciones de la osmolaridad y la propia hiperglucemia por sí misma podrían actuar como factor causal en la etiopatogenia del SDO.

