




Natalizumab es un anticuerpo monoclonal inhibidor de la migración leucocitaria a través de la barrera hematoencefálica, autorizado para el tratamiento de la esclerosis múltiple remitente-recurrente.
ObjetivoRealizar una revisión y actualización de los aspectos farmacológicos y terapéuticos de natalizumab, con especial énfasis en los datos de eficacia, efectividad y seguridad publicados más recientemente.
DesarrolloVarios ensayos clínicos aleatorizados en pacientes con formas recurrentes de esclerosis múltiple han demostrado que natalizumab reduce considerablemente la actividad clínica y radiológica de la enfermedad. El análisis post hoc de ensayos clínicos fase III y los resultados de estudios observacionales postautorización indican que natalizumab incrementa significativamente la proporción de pacientes con respuesta clínica y radiológica completa, y que es eficaz en aquellos con formas muy activas de esclerosis múltiple y con respuesta subóptima a otras terapias. Al igual que otros anticuerpos monoclonales, natalizumab puede causar reacciones de hipersensibilidad, siendo graves en el 1% de los pacientes. Otros efectos adversos de natalizumab son en general leves o poco frecuentes. No obstante, se han detectado varios casos de leucoencefalopatía multifocal progresiva en pacientes tratados con natalizumab en monoterapia. El riesgo de esta grave complicación parece incrementarse con el número de dosis recibidas.
ConclusiónNatalizumab ha demostrado una relación beneficio-riesgo favorable en el tratamiento de la esclerosis múltiple remitente-recurrente. El riesgo potencial de leucoencefalopatía multifocal progresiva, sin embargo, obliga a la selección cuidadosa de los pacientes y a seguir protocolos de actuación específicos durante su administración.
Natalizumab is a monoclonal antibody that inhibits leukocyte migration across the blood-brain barrier and has been approved for the treatment of relapsing-remitting multiple sclerosis.
ObjectiveTo provide a review and update of the pharmacological and therapeutic characteristics of natalizumab, with special emphasis on the most recently published data on the efficacy, effectiveness and safety of this drug.
DevelopmentSeveral randomized clinical trials in patients with relapsing forms of multiple sclerosis have demonstrated that natalizumab substantially reduces clinical and radiological disease activity. Post hoc analysis of phase III clinical trials and the results of post-approval observational studies indicate that natalizumab significantly increases the proportion of patients with complete clinical and radiological response and is effective in patients with highly active forms of multiple sclerosis and suboptimal response to other treatments. Like other monoclonal antibodies, natalizumab can cause hypersensitivity reactions, which are severe in 1% of patients. Other adverse effects are generally mild or infrequent. Nevertheless, several cases of progressive multifocal leukoencephalopathy have been detected in patients treated with natalizumab monotherapy. The risk of this severe complication seems to increase with the number of doses administered.
ConclusionNatalizumab has a favorable risk-benefit ratio in the treatment of relapsing -remitting multiple sclerosis. However, because of the potential risk of progressive multifocal leukoencephalopathy, patients must be carefully selected and specific protocols must be followed during the drug's administration.
La esclerosis múltiple (EM) es una enfermedad inflamatoria desmielinizante del sistema nervioso central (SNC) que afecta a unos 2,5 millones de personas en todo el mundo1. Su impacto socioeconómico es muy elevado, debido principalmente a la larga duración de la enfermedad, la mayor incidencia en adultos jóvenes y la consiguiente pérdida temprana de productividad, y el coste de los tratamientos y los cuidados multidisciplinares que los pacientes precisan2. La EM se manifiesta habitualmente en la tercera o cuarta década de la vida con un curso clínico recurrente, evolucionando posteriormente hacia una condición crónica que se caracteriza por la progresión sostenida de la discapacidad1. Estudios sobre la historia natural de la EM revelan que aproximadamente el 50% de los pacientes requiere asistencia para deambular 15 años después del diagnóstico, y la mediana de tiempo que tardan en alcanzar un grado elevado de discapacidad es de unos 30 años3.
El objetivo de cualquier terapia modificadora de la EM es reducir la frecuencia y gravedad de las recaídas, y prevenir o retrasar la evolución hacia una fase progresiva. La evidencia disponible sugiere que la EM es una enfermedad de etiología autoinmune y, de acuerdo con este concepto, su tratamiento se ha sustentado hasta ahora en el empleo de fármacos inmunomoduladores, como el interferón β (IFNβ) o el acetato de glatirámero (AG)4. A pesar del importante avance terapéutico que supuso la introducción de estos fármacos a mediados de los noventa, su eficacia es sólo parcial y sus efectos sobre el pronóstico a largo plazo es una cuestión por resolver5. Una década más tarde, la incorporación de natalizumab (Tysabri®; Biogen Idec, Inc., Elan Pharmaceuticals, Inc.) al arsenal terapéutico de la EM ha supuesto otro avance considerable. Natalizumab es un anticuerpo monoclonal que impide la migración leucocitaria a través de la barrera hematoencefálica. Este fármaco ha demostrado una buena relación beneficio-riesgo en pacientes con EM, reduciendo la frecuencia de brotes, la progresión de la discapacidad y las lesiones cuantificadas por métodos de neuroimagen. Esta revisión es una puesta al día de los aspectos farmacológicos y terapéuticos de natalizumab en el tratamiento de la EM, centrándose en los datos de eficacia clínica y seguridad publicados más recientemente.
Mecanismo de acciónLas hipótesis actuales sostienen que un suceso central en la patogenia de la EM es la activación de linfocitos T autorreactivos en la periferia que, tras proliferar y atravesar la barrera hematoencefálica, desencadenan una cascada de eventos inflamatorios en el SNC que causan finalmente desmielinización y daño axonal1. La migración de leucocitos a través de la barrera hematoencefálica requiere la interacción entre moléculas de adhesión expresadas en la superficie celular, como selectinas e integrinas, y sus receptores endoteliales. En particular, la unión de alta afinidad entre la integrina α4β1 y la molécula de adhesión de las células vasculares-1 (VCAM-1) permite a las células detenerse sobre el endotelio vascular y comenzar la migración transendotelial6.
Natalizumab, un anticuerpo monoclonal humanizado con una estructura IgG4, es un inhibidor selectivo de moléculas de adhesión que reconoce y se fija específicamente a la subunidad α4 de la integrina α4β17. Este fármaco bloquea la unión entre la VCAM-1 endotelial y la integrina α4β1 expresada en la superficie de linfocitos T activados y otros leucocitos mononucleares, impidiendo su adhesión al endotelio, la migración y el reclutamiento celular hacia el parénquima, y la subsiguiente actividad inflamatoria en el SNC8. Se ha sugerido que natalizumab también podría ejercer sus efectos inmunomoduladores al inhibir la interacción entre la integrina α4β1 y moléculas de la matriz extracelular como la fibronectina o la osteopontina, o al reducir el número de células dendríticas y la expresión de CMH clase II en los espacios perivasculares del SNC7,9.
FarmacocinéticaCuando se administran entre 1 y 3mg/kg de natalizumab a voluntarios sanos o a pacientes con EM, las concentraciones del fármaco son detectables en sangre durante 3 a 8 semanas10,11. Después de una única dosis, las concentraciones séricas máximas se alcanzan lentamente, en 1 o 2h tras la administración, a pesar de que básicamente el fármaco se distribuye en el sistema vascular y apenas difunde a los tejidos12. La concentración en sangre disminuye lentamente y tarda entre 1 y 2 semanas en reducirse a la mitad, manteniéndose una alta saturación de la integrina α4 durante ese período10,13,14. La permanencia en sangre aumenta al incrementar la dosis y, por lo tanto, se justifica la pauta seleccionada en los ensayos clínicos fase III y aprobada por las agencias reguladoras de 300mg administrados por vía intravenosa (i.v.) cada 4 semanas. Los parámetros farmacocinéticos de natalizumab son similares en pacientes con EM tras la administración en dosis repetidas de 300mg: la vida media de eliminación sérica es de 11±4 días, el aclaramiento sérico de 16±5ml/h y el volumen de distribución de 5,7±1,9 l11,12.
Aunque no hay estudios farmacocinéticos realizados en pacientes con insuficiencia hepática o renal, los datos disponibles no indican que deba modificarse la dosis o la pauta de administración de natalizumab en estos casos. Tampoco parece alterarse su farmacocinética si se administra junto con otros medicamentos11,12,15. Sin embargo, la presencia de anticuerpos antinatalizumab sí que puede aumentar 3 veces la velocidad del aclaramiento plasmático del fármaco, disminuyendo su concentración en sangre hasta valores que comprometen su eficacia terapéutica12.
Con la pauta habitual de tratamiento en pacientes con EM, la saturación de la integrina α4 es superior al 70% después de 4 semanas tras la última dosis de natalizumab. Si se suspende la administración del fármaco, sus concentraciones plasmáticas y sus efectos biológicos pueden persistir hasta 12 semanas14,16. Por este motivo, si se precisa acelerar su eliminación, debe recurrirse a medidas como la plasmaféresis. Ésta reduce las concentraciones plasmáticas de natalizumab en un 92% después de 3 recambios de 1,5 volúmenes de plasma. No obstante, la saturación de la integrina α4 puede no reducirse de un modo paralelo. Se requerirían 5 recambios de 1,5 volúmenes de plasma en días alternos para reducir las concentraciones de natalizumab por debajo de 1μg/ml y la saturación de la integrina α4 por debajo del 50% en el 95% de los pacientes14.
Ensayos clínicosLa eficacia de natalizumab en el tratamiento de la EM se ha evaluado en 6 ensayos clínicos aleatorizados, con enmascaramiento a doble ciego, controlados con placebo, que han incluido un total de 2.688 pacientes, de los que 1.567 recibieron natalizumab13,17–21. Cuatro de estos ensayos fueron fase II y 2 fueron fase III. Los pacientes evaluados estaban diagnosticados de EM remitente-recurrente (EMRR), aunque en los ensayos fase III también se aceptó el diagnóstico de EM secundariamente progresiva como criterio de inclusión.
El diseño y los resultados de los ensayos fase II se resumen en la tabla 1. El principal objetivo de 3 de estos estudios fue evaluar la eficacia de natalizumab en monoterapia o en combinación con el régimen estándar de AG en la actividad de la enfermedad determinada mediante resonancia magnética (RM) cerebral17–19. Un cuarto estudio examinó la hipótesis de que el tratamiento con natalizumab durante la fase aguda de un brote podría acelerar la recuperación clínica del paciente13. En conjunto, los resultados obtenidos aportaron, por primera vez, pruebas convincentes de que un anticuerpo monoclonal inhibidor selectivo de la integrina α4 era una alternativa potencialmente eficaz y bien tolerada en el tratamiento de la EM, lo que justificaba la realización de ensayos clínicos fase III para confirmar la eficacia clínica y seguridad de natalizumab.
Ensayos clínicos aleatorizados fase II de natalizumab para el tratamiento de la esclerosis múltiple
| Autor (año) | Estudio | Criterios de inclusión | Tratamiento | N | Resultados para los objetivos primarios |
| Tubridy et al (1999)17 | Eficacia de natalizumab en la actividad de la enfermedad evaluada mediante RM en pacientes con EMRR y EMSP | Edad 18-55 años, EDSS 2-7, ≥ 2 brotes en los 18 meses previos, > 4 semanas desde el último brote | Natalizumab 3 mg/kg i.v. o placebo (1:1), 2 dosis administradas en las semanas 0 y 4 | 72 | Media del número acumulado de nuevas lesiones activas en la RM cerebral entre las semanas 1 y 12 del estudio: 1,8 natalizumab 3 mg/kg, 3,6 placebo (p<0,042) |
| Miller et al (2003)18 | Eficacia de natalizumab en la actividad de la enfermedad evaluada mediante RM en pacientes con EMRR y EMSP | Edad 18-65 años, EDSS 2-6,5, ≥ 2 brotes en los 2 años previos, ≥ 3 lesiones en T2 en la RM cerebral | Natalizumab 3 mg/kg i.v., natalizumab 6 mg/kg i.v. o placebo (1:1:1), cada 4 semanas durante 6 meses | 213 | Media del número de nuevas lesiones Gd+ en la RM cerebral durante el período de tratamiento: 0,7 natalizumab 3 mg/kg, 1,1 natalizumab 6 mg/kg, 9,6 placebo (p<0,001) |
| O’Connor et al (2004)13 | Eficacia clínica de natalizumab en pacientes con EMRR y EMSP durante la fase aguda del brote | Edad 18-65 años, EDSS 0-5,5, estabilidad clínica durante ≥ 30 días previos, síntomas de un brote durante > 24h pero<96h, EDSS > 3 durante el brote | Natalizumab 1 mg/kg i.v., natalizumab 3 mg/kg i.v. o placebo (1:1:1), dosis única | 180 | Comparación del cambio medio en la EDSS durante la primera semana de tratamiento entre ambos grupos del estudio: no se observaron diferencias entre natalizumab y placebo |
| Goodman et al (2009)19 | Eficacia de natalizumab combinado con AG en la actividad radiológica de la enfermedad en pacientes con EMRR y EMSP | Edad 18-55 años, EDSS 0-5, tratamiento con AG durante ≥ 12 meses, ≥ 1 brote en los 12 meses previos | Natalizumab 300mg i.v. o placebo (1:1) cada 4 semanas, además de AG 20mg s.c. diario, durante ≤ 24 semanas | 110 | Tasa de formación de nuevas lesiones activas durante el período de tratamiento: 0,03 natalizumab 300mg y AG 20mg, 0,11 placebo y AG (p<0,031) |
AG: acetato de glatirámero; EDSS: escala expandida del estado de discapacidad; EMRR: esclerosis múltiple remitente-recurrente; EMSP: esclerosis múltiple secundariamente progresiva; i.v.: intravenoso; RM: resonancia magnética; s.c.: subcutáneo.
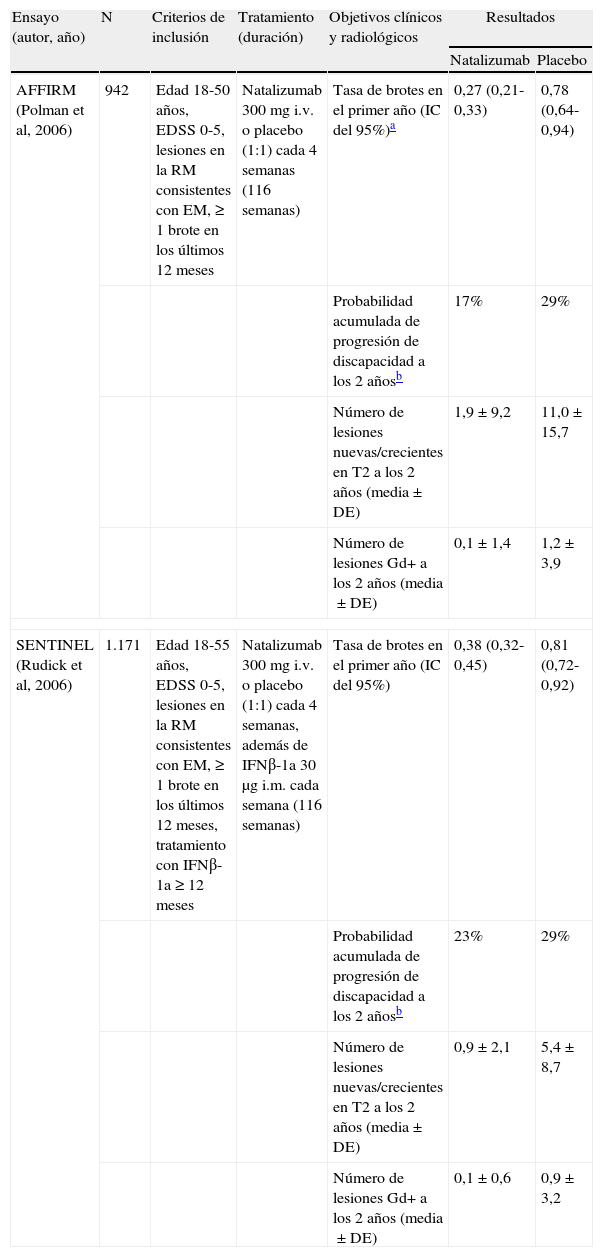
Los 2 ensayos fase III en los que natalizumab fue evaluado para el tratamiento de la EMRR, denominados con los acrónimos AFFIRM y SENTINEL, presentaban un diseño similar20,21. Ambos incluyeron pacientes con una puntuación en la escala expandida del estado de discapacidad (EDSS) de 0 a 5,0, lesiones en la RM cerebral compatibles con EM y que hubiesen presentado al menos un brote en los 12 meses previos. En el estudio SENTINEL se exigía, como criterio adicional, que los pacientes hubiesen recibido tratamiento con IFNβ-1a durante al menos 12 meses antes de la aleatorización21. En ambos ensayos se comparó la eficacia y seguridad de natalizumab 300mg administrado i.v. cada 4 semanas frente a placebo. En el estudio SENTINEL todos los participantes recibieron además tratamiento con IFNβ-1a 30μg administrado por vía intramuscular cada semana21. Los objetivos primarios se diferenciaban en 2 tiempos preestablecidos en los 2 ensayos. El primer objetivo primario fue evaluar si, tras 1 año de tratamiento, la frecuencia de brotes en el grupo de natalizumab era menor que en el grupo placebo. A los 2 años, en el momento de finalizar el tratamiento, el objetivo primario consistió en comparar entre los grupos del estudio la probabilidad acumulada de progresión sostenida de la discapacidad, definida como un incremento de al menos 1,0 puntos en la escala EDSS mantenido durante 12 semanas para los pacientes con una puntuación basal ≥ 1,0, o un incremento sostenido de al menos 1,5 puntos durante 12 semanas cuando la puntuación basal era igual a 0.
Los resultados de ambos ensayos se resumen en la tabla 2. Se había planificado previamente un análisis intermedio del primer objetivo cuando se dispusiese de los datos de los primeros 900 pacientes-año evaluados en el estudio AFFIRM y 1.200 pacientes-año en el estudio SENTINEL20,21. En ese momento, los pacientes tratados con natalizumab en los 2 ensayos presentaban una disminución relativa en la frecuencia anual de recaídas frente a placebo del 68 y el 54%, respectivamente (p<0,001). Esta diferencia se mantuvo cuando se analizó el total de los datos del primer año (el 65 y el 53%; p<0,001) y al finalizar el segundo año del estudio (el 68 y el 55%; p ≤ 0,001). La frecuencia de recaídas fue similar en ambos ensayos, a pesar de que en el estudio SENTINEL todos los participantes recibieron IFNβ-1a. La proporción de pacientes libres de brotes durante los 2 años en los estudios AFFIRM y SENTINEL fue del 67 y el 54% en los grupos de natalizumab, frente al 41 y el 32% en los grupos placebo, respectivamente (p<0,001). El segundo objetivo primario, la probabilidad acumulada de progresión de la discapacidad a los 2 años, también fue significativamente menor en los grupos de natalizumab, siendo los valores del cociente de riesgo de 0,58 y 0,76, lo que supone una disminución del riesgo de progresión de discapacidad frente a placebo del 42 y el 24%20,21.
Ensayos clínicos aleatorizados fase III de natalizumab para el tratamiento de la esclerosis múltiple
| Ensayo (autor, año) | N | Criterios de inclusión | Tratamiento (duración) | Objetivos clínicos y radiológicos | Resultados | |
| Natalizumab | Placebo | |||||
| AFFIRM (Polman et al, 2006) | 942 | Edad 18-50 años, EDSS 0-5, lesiones en la RM consistentes con EM, ≥ 1 brote en los últimos 12 meses | Natalizumab 300mg i.v. o placebo (1:1) cada 4 semanas (116 semanas) | Tasa de brotes en el primer año (IC del 95%)a | 0,27 (0,21-0,33) | 0,78 (0,64-0,94) |
| Probabilidad acumulada de progresión de discapacidad a los 2 añosb | 17% | 29% | ||||
| Número de lesiones nuevas/crecientes en T2 a los 2 años (media±DE) | 1,9±9,2 | 11,0±15,7 | ||||
| Número de lesiones Gd+ a los 2 años (media±DE) | 0,1±1,4 | 1,2±3,9 | ||||
| SENTINEL (Rudick et al, 2006) | 1.171 | Edad 18-55 años, EDSS 0-5, lesiones en la RM consistentes con EM, ≥ 1 brote en los últimos 12 meses, tratamiento con IFNβ-1a ≥ 12 meses | Natalizumab 300mg i.v. o placebo (1:1) cada 4 semanas, además de IFNβ-1a 30μg i.m. cada semana (116 semanas) | Tasa de brotes en el primer año (IC del 95%) | 0,38 (0,32-0,45) | 0,81 (0,72-0,92) |
| Probabilidad acumulada de progresión de discapacidad a los 2 añosb | 23% | 29% | ||||
| Número de lesiones nuevas/crecientes en T2 a los 2 años (media±DE) | 0,9±2,1 | 5,4±8,7 | ||||
| Número de lesiones Gd+ a los 2 años (media±DE) | 0,1±0,6 | 0,9±3,2 | ||||
DE: desviación estándar; EDSS: escala expandida del estado de discapacidad; EMRR: esclerosis múltiple remitente-recurrente; IC: intervalo de confianza; IFNβ-1a: interferón β-1a; i.v.: i.m.: intramuscular; intravenoso; RM: resonancia magnética.
p<0,001 para todas las comparaciones excepto probabilidad acumulada de progresión de discapacidad a los 2 años en el estudio SENTINEL (p=0,02).
Aunque los resultados de los hallazgos de la RM cerebral constituían un objetivo secundario, en ambos ensayos natalizumab redujo el número de lesiones nuevas o crecientes hiperintensas en secuencias ponderadas en T2 en un 83% frente a placebo (p<0,001), y entre el 89 y el 92% el número de lesiones captadoras de gadolinio en secuencias ponderadas en T1 (p<0,001) después de 2 años de tratamiento20,21. En el estudio AFFIRM, el tratamiento con natalizumab se asoció además con una disminución del 76% en el número de lesiones nuevas hipointensas en secuencias ponderadas en T1 respecto al grupo placebo (p<0,001)22. Asimismo, el volumen de lesiones en secuencias ponderadas en T2 se redujo en un 9,4% en el grupo de natalizumab y se incrementó en un 8,8% en el grupo placebo (p<0,001)22. Estos datos sugieren un efecto sostenido y relevante del tratamiento con natalizumab en la prevención de la aparición de nuevas lesiones en pacientes con EMRR.
Un análisis post hoc en pacientes del estudio AFFIRM evaluó la capacidad de natalizumab para alcanzar una respuesta clínica y radiológica completa23. La ausencia de actividad clínica de la enfermedad se definió como ausencia de brotes y de progresión de la discapacidad mantenida a las 12 semanas. La ausencia de actividad radiológica se definió como la ausencia de lesiones captadoras de gadolinio en secuencias ponderadas en T1 y de lesiones nuevas o crecientes hiperintensas en secuencias ponderadas en T2 de RM. La ausencia de actividad de la enfermedad se definió como la ausencia de ambas variables. Una proporción significativamente mayor de pacientes tratados con natalizumab se halló libre de actividad clínica (el 64 frente al 39%; p<0,0001), libre de actividad radiológica (el 58 frente al 14%; p<0,0001) y libre de actividad de la enfermedad (el 37 frente al 7%; p<0,0001) en comparación con el grupo placebo durante los 2 años de duración del ensayo23.
Otro estudio llevó a cabo un análisis post hoc de la respuesta al tratamiento con natalizumab en los pacientes de los estudios AFFIRM y SENTINEL con EMRR muy activa, definida por 2 o más brotes en el año anterior al estudio y la presencia de al menos 1 lesión captadora de gadolinio en secuencias ponderadas en T1 en una RM previa a la aleatorización24. En los pacientes con EMRR muy activa del estudio AFFIRM, natalizumab redujo el riesgo acumulado de progresión de la discapacidad confirmada a las 24 semanas en un 64% frente a placebo (p=0,008), y la tasa anualizada de brotes en un 81% (p<0,001), durante los 2 años de tratamiento. En el estudio SENTINEL, la terapia combinada con natalizumab e IFNβ-1a también redujo el riesgo acumulado de progresión de la discapacidad confirmada a las 24 semanas en un 58% frente a IFNβ-1a y placebo (p=0,038), y la tasa anualizada de brotes en un 76% (p<0,001) en el mismo tipo de pacientes24. A pesar de que se trata de un análisis de subgrupos, los datos de este estudio sugieren que natalizumab es eficaz en formas de la enfermedad con elevada actividad.
Se han publicado análisis a posteriori del efecto de natalizumab sobre otros objetivos terciaros preestablecidos. La función visual, no relacionada específicamente con los episodios de neuritis óptica, es una importante medida de la discapacidad en pacientes con EM. En los estudios AFFIRM y SENTINEL se examinó la agudeza visual mediante el optotipo de Sloan cada 12 semanas. Natalizumab redujo el riesgo de pérdida clínicamente relevante de visión de bajo contraste (predefinida como una reducción de 2 líneas en el optotipo) en un 28 y un 35%, respectivamente, frente a los grupos controles25.
El análisis de la calidad de vida relacionada con la salud (CVRS) aporta una valoración subjetiva y global del efecto de un tratamiento sobre el estado de la salud del paciente. En los estudios AFFIRM y SENTINEL se llevó a cabo un análisis riguroso de la CVRS mediante el cuestionario de salud SF-36 y una escala analógica visual (EAV) de bienestar global, que se evaluaron antes de iniciar el tratamiento y en las semanas 24, 52 y 10426. En el estudio AFFIRM, el tratamiento con natalizumab se asoció con una mejoría en las dimensiones de función física y salud mental del cuestionario SF-36 en la semana 104 respecto a la situación basal, en comparación con el grupo placebo. También se observó una mejoría en la dimensión de función física en el resto de los tiempos evaluados. En el estudio SENTINEL, este parámetro también fue significativamente mejor en las semanas 52 y 104 en el grupo de natalizumab. Asimismo, los cambios en la EAV fueron favorables para natalizumab en el estudio AFFIRM. Estos datos sugieren que el tratamiento con este fármaco podría asociarse con una mejoría en la CVRS de los pacientes con EM26.
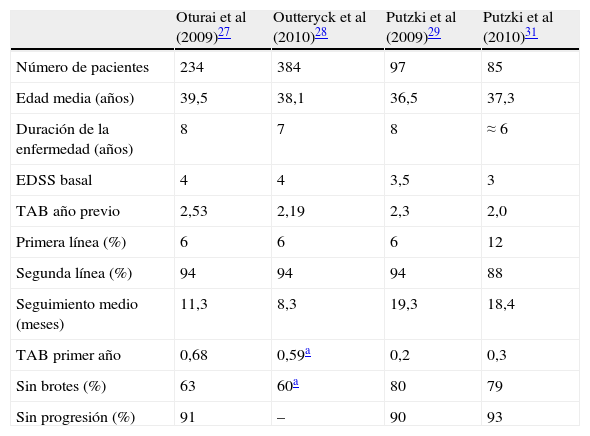
Estudios observacionalesLos ensayos clínicos fase III demostraron la considerable eficacia de natalizumab en la reducción de la actividad clínica y radiológica en pacientes con EMRR. No obstante, debido a informes que notificaban la aparición de casos de leucoencefalopatía multifocal progresiva (LMP) en pacientes tratados con natalizumab, las indicaciones terapéuticas aprobadas por la European Medicines Agency (EMA) restringen su uso a pacientes con EMRR con elevada actividad a pesar del tratamiento con IFNβ y con formas de EMRR grave de evolución rápida. Ningún ensayo clínico ha evaluado específicamente la eficacia de natalizumab como terapia de segunda línea en pacientes con respuesta subóptima a IFNβ o AG, ni en formas agresivas de EMRR. En este sentido, los estudios observacionales postautorización realizados en varios centros europeos aportan información relevante respecto al uso de natalizumab en las condiciones autorizadas27–31. Estos indican que natalizumab se utiliza como segunda línea de tratamiento entre el 88 y el 94% de los pacientes. En conjunto, y a pesar de sus limitaciones metodológicas, los datos publicados sugieren que natalizumab es efectivo en pacientes con respuesta subóptima a otros fármacos durante al menos el primer año de tratamiento, con un grado de beneficio terapéutico similar al observado en los ensayos clínicos aleatorizados (tabla 3)27–29,31.
Estudios observacionales postautorización de natalizumab para el tratamiento de la esclerosis múltiple
| Oturai et al (2009)27 | Outteryck et al (2010)28 | Putzki et al (2009)29 | Putzki et al (2010)31 | |
| Número de pacientes | 234 | 384 | 97 | 85 |
| Edad media (años) | 39,5 | 38,1 | 36,5 | 37,3 |
| Duración de la enfermedad (años) | 8 | 7 | 8 | ≈ 6 |
| EDSS basal | 4 | 4 | 3,5 | 3 |
| TAB año previo | 2,53 | 2,19 | 2,3 | 2,0 |
| Primera línea (%) | 6 | 6 | 6 | 12 |
| Segunda línea (%) | 94 | 94 | 94 | 88 |
| Seguimiento medio (meses) | 11,3 | 8,3 | 19,3 | 18,4 |
| TAB primer año | 0,68 | 0,59a | 0,2 | 0,3 |
| Sin brotes (%) | 63 | 60a | 80 | 79 |
| Sin progresión (%) | 91 | – | 90 | 93 |
EDSS: escala expandida del estado de discapacidad; TAB: tasa anualizada de brotes.
Primera línea: porcentaje de pacientes tratados con natalizumab como primera opción terapéutica; segunda línea: porcentaje de pacientes tratados previamente con otros fármacos modificadores de la enfermedad.
De forma similar a otros agentes terapéuticos péptidicos, natalizumab puede inducir la formación de anticuerpos antinatalizumab durante su administración. En los estudios AFFIRM y SENTINEL se determinó la presencia de anticuerpos antinatalizumab mediante ELISA cada 12 semanas durante los 2 años de tratamiento32. Se definió como paciente persistentemente positivo aquel con una concentración de anticuerpos ≥ 0,5μg/ml en 2 o más determinaciones realizadas con un intervalo mínimo de 6 semanas. En el estudio AFFIRM, natalizumab indujo la formación de anticuerpos en el 9% de los pacientes: el 6% fue persistentemente positivo y el 3% transitoriamente positivo (una única determinación positiva). En el estudio SENTINEL, el 6% de los pacientes fue persistentemente positivo y el 5% transitoriamente positivo. En la mayor parte de los casos (88-96%), los anticuerpos se detectaron por primera vez dentro de las primeras 12 semanas de tratamiento. En ambos ensayos, la existencia de anticuerpos antinatalizumab se relacionó con una disminución en la concentración sérica de natalizumab. La presencia de anticuerpos persistentes se asoció con una reducción en la eficacia clínica y radiológica de natalizumab. En los pacientes transitoriamente positivos, la eficacia completa de natalizumab se alcanzó después de, aproximadamente, 6 meses desde el inicio del tratamiento, momento en el que los anticuerpos se negativizaron32.
Tolerancia y seguridadEn general, natalizumab fue bien tolerado en los ensayos clínicos. En 1617 pacientes con EM tratados con natalizumab en ensayos clínicos controlados con placebo, durante un máximo de dos años, se produjeron acontecimientos adversos que motivaron la retirada del tratamiento en el 5,8% de los pacientes tratados con natalizumab y en el 4,8% de los tratados con placebo. Las reacciones de hipersensibilidad fueron el motivo más frecuente de suspensión del tratamiento12. El 43,5% de los pacientes tratados con natalizumab comunicó reacciones farmacológicas adversas (placebo 39,6%). Los acontecimientos adversos comunicados con natalizumab con una incidencia 0,5% superior a placebo fueron: cefalea, mareos, vómitos, náuseas, artralgias, infección del tracto urinario, nasofaringitis, temblores, fiebre, fatiga, urticaria y reacciones de hipersensibilidad33.
Reacciones a la perfusiónEn los ensayos clínicos controlados con placebo se definió reacción a la perfusión como cualquier acontecimiento adverso ocurrido durante la perfusión de 1h de duración o en el curso de la hora siguiente. Estas reacciones se observaron en el 24% de los pacientes tratados con natalizumab en monoterapia y en el 18% de los que recibieron placebo (p=0,04)20. La mayor parte de estas reacciones sólo requirió tratamiento sintomático y no condujo a la discontinuación del tratamiento, siendo graves en menos del 1% de los casos15,20. Las reacciones a la perfusión más frecuentes comunicadas en pacientes tratados con natalizumab fueron cefalea, mareo, náuseas, temblores y urticaria12.
Reacciones de hipersensibilidadEn el estudio AFFIRM, los acontecimientos adversos notificados como reacciones de hipersensibilidad sucedieron en el 4% de los pacientes tratados con natalizumab. Cinco (0,8%) de las reacciones fueron consideradas sistémicas graves (reacciones anafilácticas o anafilactoides)20. Las reacciones de hipersensibilidad se producen normalmente durante la perfusión de natalizumab o durante la hora siguiente, y suelen manifestarse en forma de urticaria acompañada o no de otros síntomas. El riesgo de presentar estas reacciones es mayor durante los primeros meses de tratamiento, siendo el período de mayor riesgo entre la primera y la séptima perfusiones, sobre todo durante la segunda33,34. También parece incrementarse el riesgo en pacientes expuestos nuevamente a natalizumab después de una breve exposición inicial (1 o 2 dosis) y un período prolongado (3 o más meses) sin tratamiento33. Cuando se presentan, este tipo de reacciones parecen responder adecuadamente al tratamiento con adrenalina, antihistamínicos y corticosteroides34. Debe suspenderse el fármaco permanentemente en los pacientes que hayan presentado una reacción de hipersensibilidad33.
En los ensayos clínicos fase III se detectó una mayor incidencia de reacciones a la perfusión y de hipersensibilidad en pacientes con anticuerpos antinatalizumab persistentemente positivos. En el estudio AFFIRM, las reacciones de hipersensibilidad sucedieron en el 46% de los pacientes con anticuerpos persistentemente positivos, en el 15% de los pacientes con anticuerpos transitoriamente positivos y en el 0,7% de los pacientes sin anticuerpos. Sin embargo, no puede predecirse qué pacientes con anticuerpos persistentemente positivos desarrollarán una reacción de hipersensibilidad.
Aunque la mayor parte de reacciones de hipersensibilidad sucede en las primeras 2h tras la perfusión de natalizumab, también pueden ocurrir reacciones de hipersensibilidad diferida desde algunas horas hasta varios días después de la administración35–37. Estas reacciones también suelen suceder durante los primeros meses de tratamiento, y son clínicamente similares a la enfermedad del suero, por lo que se postula un mecanismo de hipersensibilidad tipo III. Los síntomas habituales son fiebre, cefalea, cervicalgia, prurito, malestar general y artralgias. En pacientes con reacciones diferidas y anticuerpos antinatalizumab sería necesario suspender el tratamiento. En opinión de algunos autores, si no se detectan anticuerpos, el pretratamiento con antihistamínicos y corticosteroides y la reducción de la velocidad de perfusión podrían ser suficientes para controlar los síntomas36,38.
Alteración de las pruebas de laboratorioEl tratamiento con natalizumab se asocia con un incremento en las concentraciones plasmáticas de linfocitos, monocitos, eosinófilos y basófilos. Éste es un efecto consistente con el mecanismo de acción de natalizumab, que reduce la migración celular fuera del torrente circulatorio. De hecho, no se observan incrementos de los neutrófilos, que no expresan integrina α4. Este efecto no parece asociarse con ninguna manifestación clínica y es reversible tras la retirada del tratamiento33.
En los ensayos clínicos fase III no se observaron diferencias significativas en la elevación de enzimas hepáticas entre los grupos de natalizumab y el grupo placebo. Sin embargo, con posterioridad se han comunicado varios casos de daño hepático clínicamente relevante en pacientes tratados con natalizumab, apareciendo de nuevo en algunos de ellos tras la readministración. La EMA tiene constancia de al menos 29 casos de daño hepático, de los que dos tercios se consideraron graves. Esta agencia recomienda la monitorización continuada de la función hepática y suspender el tratamiento en caso de que se presenten reacciones significativas39.
NeoplasiasNo se detectaron diferencias apreciables en la incidencia de neoplasias en pacientes tratados con natalizumab respecto a los que recibieron placebo en los ensayos clínicos aleatorizados. Además, la incidencia de las neoplasias observadas, como cáncer de mama o carcinoma basocelular, no excedió la esperada en la población general. En el estudio AFFIRM, un paciente con antecedente de melanoma maligno falleció debido a un melanoma mestastásico20. En la fase postautorización se notificaron 2 casos más de melanoma en pacientes tratados con natalizumab40. También se ha comunicado un caso de linfoma cerebral primario en un paciente de 40 años que había sido tratado previamente con azatioprina, después de recibir 21 dosis de natalizumab41. Hay otra notificación de una paciente de 30 años en la que el tratamiento con natalizumab podría haber influido en el crecimiento de un linfoma cerebral primario42. No obstante, los datos disponibles actualmente son insuficientes para determinar si el fármaco puede incrementar el riesgo de melanoma o linfoma cerebral primario, o modificar su evolución.
InfeccionesSegún datos provenientes de los ensayos clínicos fase III, natalizumab no incrementa el riesgo global de infecciones respecto a placebo. En estos estudios, la tasa de infecciones fue de aproximadamente 1,5 por paciente-año en ambos grupos, y la naturaleza de las infecciones fue similar. Se observó un discreto incremento, no significativo, en la incidencia de neumonía e infecciones del tracto urinario en los pacientes que recibieron natalizumab. Entre estos también se detectó un caso de diarrea por Cryptosporidium, una infección oportunista. Excepto la LMP, no se observaron otras infecciones oportunistas durante los ensayos clínicos en pacientes con EM. Recientemente se ha publicado un caso de toxoplasmosis ocular en un paciente de 28 años y un caso de candidiasis cutánea grave en una mujer de 61 años, ambos después de la undécima dosis de natalizumab43,44.
En los ensayos clínicos se produjeron infecciones por virus varicela-zóster y virus del herpes simple con una frecuencia ligeramente mayor en pacientes tratados con natalizumab respecto a placebo. En la experiencia postautorización se han descrito casos graves, incluyendo un caso de meningitis herpética y otro caso mortal de encefalitis herpética12. Por tanto, aunque no hay una relación demostrada entre el riesgo de infecciones oportunistas o por virus herpes y natalizumab, es recomendable vigilar cuidadosamente la aparición de estas infecciones en los pacientes tratados.
Leucoencefalopatía multifocal progresivaEl estudio SENTINEL se suspendió en febrero de 2005, poco tiempo antes de concluir, debido a 2 notificaciones de LMP en pacientes que habían recibido natalizumab en combinación con IFNβ-1a45,46. Posteriormente se conoció otro caso de LMP en un paciente tratado con natalizumab para la enfermedad de Crohn47. Estos hechos condujeron a la retirada del fármaco por el laboratorio responsable de su comercialización, que había sido autorizada 3 meses antes en Estados Unidos48. Se llevó a cabo entonces una revisión exhaustiva de los pacientes tratados con natalizumab hasta ese momento, para descartar posibles casos adicionales de LMP. El análisis incluyó 3.389 participantes en ensayos clínicos, de los cuales 3.117 recibieron natalizumab y 273 placebo. También se contactó con los médicos que habían administrado natalizumab a cerca de 7.000 pacientes entre noviembre de 2004 y febrero de 2005 tras la comercialización del producto. No se detectaron nuevos casos, y se estimó que la incidencia de LMP era de 1 caso por cada 1.000 pacientes expuestos a una media de 18 dosis mensuales de natalizumab49. Basándose en estos resultados, en junio de 2006 se autorizó la reintroducción de natalizumab para el tratamiento de la EMRR en Estados Unidos y Europa.
Desde junio de 2006 hasta enero de 2010, cerca de 66.000 pacientes han recibido al menos 1 dosis de natalizumab en todo el mundo. En ese período se han confirmados 31 nuevos casos de LMP asociada al uso de natalizumab en monoterapia en Estados Unidos y Europa, de los que 23 han sucedido en pacientes tratados durante más de 2 años50,51. El análisis de los datos disponibles sugiere que el riesgo de LMP se incrementa al aumentar el número de dosis recibidas y que la tasa acumulada de LMP es de 0,8 casos por cada 1.000 pacientes tratados con 12 o más dosis de natalizumab, y de 1,3 casos por cada 1.000 pacientes tratados con 24 o más dosis51. La magnitud del riesgo en pacientes que han recibido 36 o más dosis no puede establecerse en la actualidad.
La LMP es una enfermedad desmielinizante del SNC causada por una infección lítica de los oligodendrocitos por el virus JC, un poliomavirus ubicuo en la población humana. En países desarrollados, del 70 al 90% de los adultos tiene anticuerpos detectables contra el virus JC. La primoinfección ocurre a edades tempranas y es típicamente asintomática, aunque los mecanismos de transmisión no se conocen bien52,53. Posteriormente, el virus permanece quiescente en los riñones, órganos linfoides y posiblemente en el SNC54,55. La LMP aparece característicamente en situaciones de compromiso inmunitario y se postula que sucede como consecuencia de la reactivación del virus JC latente o por una mutación adaptativa que favorece la infección del SNC53,56,57.
La LMP se caracteriza clínicamente por déficits neurológicos de instauración subaguda, incluyendo trastornos cognitivos, visuales y motores. La RM cerebral muestra habitualmente lesiones bilaterales y asimétricas subcorticales en tronco del encéfalo y cerebelo, hipointensas en secuencias ponderadas en T2 y FLAIR, sin realce tras la administración de contraste ni efecto masa significativo. En un contexto clínico y radiológico apropiado, la detección del ADN del virus JC mediante PCR en el líquido cefalorraquídeo, con una sensibilidad aproximada del 80% y una especificidad superior al 95%, respalda claramente el diagnóstico54,56,58. Esta prueba puede ser negativa inicialmente, y en caso de sospecha diagnóstica se recomienda repetir el estudio posteriormente53,59.
Se desconocen las bases de la relación causal entre la exposición a natalizumab y la LMP. Por una parte, el efecto antimigratorio de natalizumab sobre los linfocitos T podría interferir con los mecanismos de vigilancia inmune en el SNC, permitiendo así la infección por el virus JC60; por otra, natalizumab incrementa en la circulación sanguínea el número de linfocitos B y pre-B61, y de células progenitoras hematopoyéticas CD34+62,63, que podrían actuar como reservorio del virus JC y facilitar su diseminación hacia el SNC53,64–66.
Actualmente, no hay ningún método que permita estimar el riesgo individual de LMP o predecir su desarrollo66,67. Por lo tanto, es crucial una vigilancia clínica estrecha de los pacientes con EM en tratamiento con natalizumab para detectar precozmente la posible aparición de LMP. Además, se debe disponer de un estudio de RM cerebral de referencia previo al inicio del tratamiento y repetirlo anualmente33,50. Por otra parte, la EM y la LMP son enfermedades que causan alteraciones clínicas y radiológicas que pueden ser similares, y se ha elaborado un algoritmo detallado que orienta en la evaluación diagnóstica de los pacientes con EM tratados con natalizumab que presentan síntomas neurológicos nuevos o un empeoramiento de los existentes (referencias 16 y 59).
No hay un tratamiento con eficacia demostrada para la LMP, y la enfermedad suele progresar hasta la muerte, siendo la supervivencia media de 6 meses68. La experiencia previa indica que la reconstitución inmunitaria se asocia a un mejor pronóstico54,69. Por lo tanto, éste se considera un objetivo prioritario del tratamiento de la LMP, y en pacientes que reciben natalizumab es fundamental retirar el fármaco en caso de que se sospeche esta enfermedad. Como los efectos biológicos de natalizumab pueden prolongarse meses, un método para acelerar su eliminación puede ser la plasmaféresis14. No obstante, como se ha explicado antes, la eficacia de este procedimiento puede verse limitada debido a la elevada afinidad de natalizumab por la integrina α4, que podría resultar en una inmunosupresión mantenida a pesar de eliminar el fármaco de la circulación.
Es importante señalar que la discontinuación del natalizumab en pacientes con EM y LMP puede asociarse a un síndrome inflamatorio de reconstitución inmune (IRIS), que puede causar un grave empeoramiento del estado del paciente45,51,70. Según datos recientes, muchos pacientes que suspendieron natalizumab debido a LMP, y que fueron sometidos a plasmaféresis o inmunoadsorción, desarrollaron un IRIS de días a semanas tras el procedimiento51. En estos casos puede considerarse el tratamiento con corticosteroides para favorecer la recuperación del paciente70,71.
Efecto de la interrupción del tratamientoEn el estudio AFFIRM se llevó a cabo un análisis de la actividad de la enfermedad después de la discontinuación del tratamiento en 51 pacientes tratados con natalizumab y 27 que recibieron placebo. En el grupo de natalizumab, la actividad de la enfermedad regresó a niveles equiparables al período pretratamiento, pero no hubo un efecto rebote entendido como un incremento en la actividad de la enfermedad respecto a dicho período. Además, los pacientes tratados con natalizumab no empeoraron más que los que recibieron placebo20. Un estudio posterior investigó la actividad de la enfermedad tras la suspensión del tratamiento en 23 participantes de los estudios AFFIRM y SENTINEL que habían recibido una mediana de 30 dosis de natalizumab, observando estabilidad clínica y radiológica durante 14 meses de seguimiento60,72. Finalmente, datos recientes del seguimiento de 946 participantes en ensayos clínicos respaldan la idea de que, tras la suspensión del tratamiento prolongado con natalizumab, la actividad de la enfermedad regresa a niveles pretratamiento pero no superiores después de 4 meses73.
Sin embargo, otro estudio ha sugerido la existencia de un efecto de rebote radiológico, definido como un incremento en el número de lesiones activas en secuencias ponderadas en T2 respecto al período pretratamiento74. Para algunos autores, esta discrepancia podría deberse a que, en dicho estudio, el efecto rebote sucedió principalmente a expensas de los pacientes que habían recibido pautas cortas de tratamiento (media de 2 dosis)38,75. No obstante, se ha descrito recientemente una serie de 7 pacientes que habían recibido entre 7 y 14 dosis de natalizumab en los que, 3 meses tras la retirada del tratamiento, se produjo una alteración del estado mental o fatiga, sin síntomas focales y múltiples lesiones captadoras de gadolinio en la RM cerebral (media: 14; rango: 8 a 21). El tratamiento con corticosteroides resultó en la resolución clínica y radiológica. Los autores sugieren que este fenómeno podría deberse a la restitución del tráfico linfocitario en el SNC, y proponen el nombre de CIRIS (CNS IRIS)76. Por lo tanto, se requieren más estudios para establecer el riesgo de un posible efecto rebote tras la retirada del tratamiento con natalizumab y su relación con el número de dosis administradas.
ConclusiónNatalizumab es el primer anticuerpo monoclonal y el primer inhibidor selectivo de la migración leucocitaria disponible para el tratamiento de la EM. Varios ensayos clínicos aleatorizados y controlados con placebo han demostrado que natalizumab reduce de forma considerable la actividad de la enfermedad y mejora los parámetros de severidad en pacientes con EMRR. Estudios adicionales sugieren que este fármaco es efectivo en el tratamiento de los pacientes con respuesta subóptima a otros tratamientos inmunomoduladores, y en pacientes con formas muy activas de EMRR. Sería de gran interés disponer de datos comparativos de la eficacia de natalizumab frente a las otras alternativas terapéuticas disponibles, con el fin de poder valorar la eficacia y el riesgo en términos relativos. No obstante, en ausencia de estos estudios, los datos disponibles son muy favorables para natalizumab. Como sucede con otros agentes peptídicos recombinantes, natalizumab puede inducir la formación de anticuerpos específicos persistentes. Esta reacción inmunogénica se asocia con una pérdida de la eficacia del fármaco e incrementa el riesgo de reacciones de hipersensibilidad. La incidencia de otros efectos adversos es baja. Sin embargo, la aparición de varios casos de LMP en pacientes tratados con natalizumab y otros anticuerpos monoclonales, indica que el desarrollo de nuevos fármacos con mecanismos de acción más selectivos, y posiblemente más eficaces, puede acompañarse también de un incremento en el riesgo de efectos adversos potencialmente graves, que obligan a la selección cuidadosa de los pacientes y a seguir protocolos de actuación específicos.
FinanciaciónA. Horga disfruta de una ayuda posformación sanitaria especializada del Instituto de Salud Carlos III.
Conflicto de interesesA. Horga declara no tener ningún conflicto de intereses. M. Tintoré ha participado en comités de asesoramiento científico para Teva Pharmaceutical Industries Ltd., Novartis y Sanofi-Aventis, y ha recibido ayudas para viajes y honorarios por conferencias de Teva Pharmaceutical Industries Ltd., Novartis, Sanofi-Aventis, Bayer Schering Pharma, Merck Serono y Biogen Idec.

