



El síndrome de Guillain-Barré (SGB) es un trastorno agudo e inmuno-mediado del sistema nervioso periférico. Sus dos subtipos básicos son el desmielinizante (AIDP) y el axonal (AMAN). El objetivo de este trabajo es abordar los mecanismos de daño axonal en la fase precoz del síndrome (≤10días del inicio sintomático).
DesarrolloSe han revisado aspectos histológicos, neurofisiológicos y de imagen descritos tanto en la AIDP como en la AMAN. Los hallazgos en la patología humana han sido contrastados con lo reportado en la neuritis alérgica experimental inducida por el componente P2 de la mielina. El edema inflamatorio de las raíces raquídeas y de los nervios espinales constituye la lesión inicial en el SGB. En los nervios espinales de casos fatales de AIDP se ha demostrado la presencia de lesiones isquémicas endoneurales, lo cual sugiere que la inflamación puede condicionar un incremento de su presión con reducción del flujo sanguíneo transperineural, que puede desencadenar fallo de la conducción y eventualmente degeneración axonal secundaria. En la AMAN con anticuerpos antigangliósido el bloqueo de la conducción por disfunción de los canales del sodio nodales puede afectar a troncos nerviosos proximales, intermedios y distales. Además de los mecanismos que operan en la AIDP, la degeneración axonal activa en la AMAN puede ir ligada a la disrupción del axolema nodal inducida por los anticuerpos anti-gangliósido.
ConclusiónEn la fase precoz del SGB hay edema inflamatorio de los troncos nerviosos proximales, que puede condicionar fallo de la conducción nerviosa y degeneración axonal activa.
Guillain-Barré syndrome (GBS) is an acute-onset, immune-mediated disease of the peripheral nervous system. It may be classified into 2 main subtypes: demyelinating (AIDP) and axonal (AMAN). This study aims to analyse the mechanisms of axonal damage in the early stages of GBS (within 10days of onset).
DevelopmentWe analysed histological, electrophysiological, and imaging findings from patients with AIDP and AMAN, and compared them to those of an animal model of myelin P2 protein-induced experimental allergic neuritis. Inflammatory oedema of the spinal nerve roots and spinal nerves is the initial lesion in GBS. The spinal nerves of patients with fatal AIDP may show ischaemic lesions in the endoneurium, which suggests that endoneurial inflammation may increase endoneurial fluid pressure, reducing transperineurial blood flow, potentially leading to conduction failure and eventually to axonal degeneration. In patients with AMAN associated with anti-ganglioside antibodies, nerve conduction block secondary to nodal sodium channel dysfunction may affect the proximal, intermediate, and distal nerve trunks. In addition to the mechanisms involved in AIDP, active axonal degeneration in AMAN may be associated with nodal axolemma disruption caused by anti-ganglioside antibodies.
ConclusionInflammatory oedema of the proximal nerve trunks can be observed in early stages of GBS, and it may cause nerve conduction failure and active axonal degeneration.
En la Conferencia Barraquer de la LIV Reunión Anual de la SEN (Barcelona, 2002) nos ocupamos de la compleja fisiopatología de la degeneración axonal en el síndrome de Guillain-Barré (SGB)1. El objetivo de esta revisión es el de actualizar la evolución de los conocimientos al respecto en el último quindenio, centrándonos en la fase precoz del curso clínico, convencionalmente establecida hasta el décimo día de evolución2.
El SGB engloba tres patrones básicos3-7: a)el desmielinizante clásico, conocido con el acrónimo anglosajón de AIDP (acute inflammatory demyelinating polyneuropathy); b)el axonal, que engloba AMAN (acute motor axonal neuropathy) y AMSAN (acute motor and sensory axonal neuropathy), y c)el síndrome de Miller Fisher (SMF). El marco clínico de la AIDP/AMAN-AMSAN es, en lo fundamental, una parálisis aguda, ascendente, flácida y arrefléxica, que alcanza su acmé dentro de las cuatro primeras semanas de curso clínico.
Con una incidencia anual entre 1 y 2 casos/100.000 habitantes, conviene recordar que las formas axonales son más prevalentes en ciertos países de Asia, mientras que en Europa y Estados Unidos AIDP es la forma predominante8-10.
Patología en la fase precoz del síndrome de Guillain-Barré: anotaciones históricas imprescindiblesEl primer estudio autópsico en la fase precoz del SGB lo efectuaron Haymaker y Kernohan11 en 50 casos, 32 de los cuales habían fallecido entre los días 2 y 10 del inicio sintomático. En los primeros cuatro días de evolución la única lesión observada fue edema endoneural, que predominaba donde las raíces raquídeas anteriores y posteriores se unen para formar los nervios espinales.
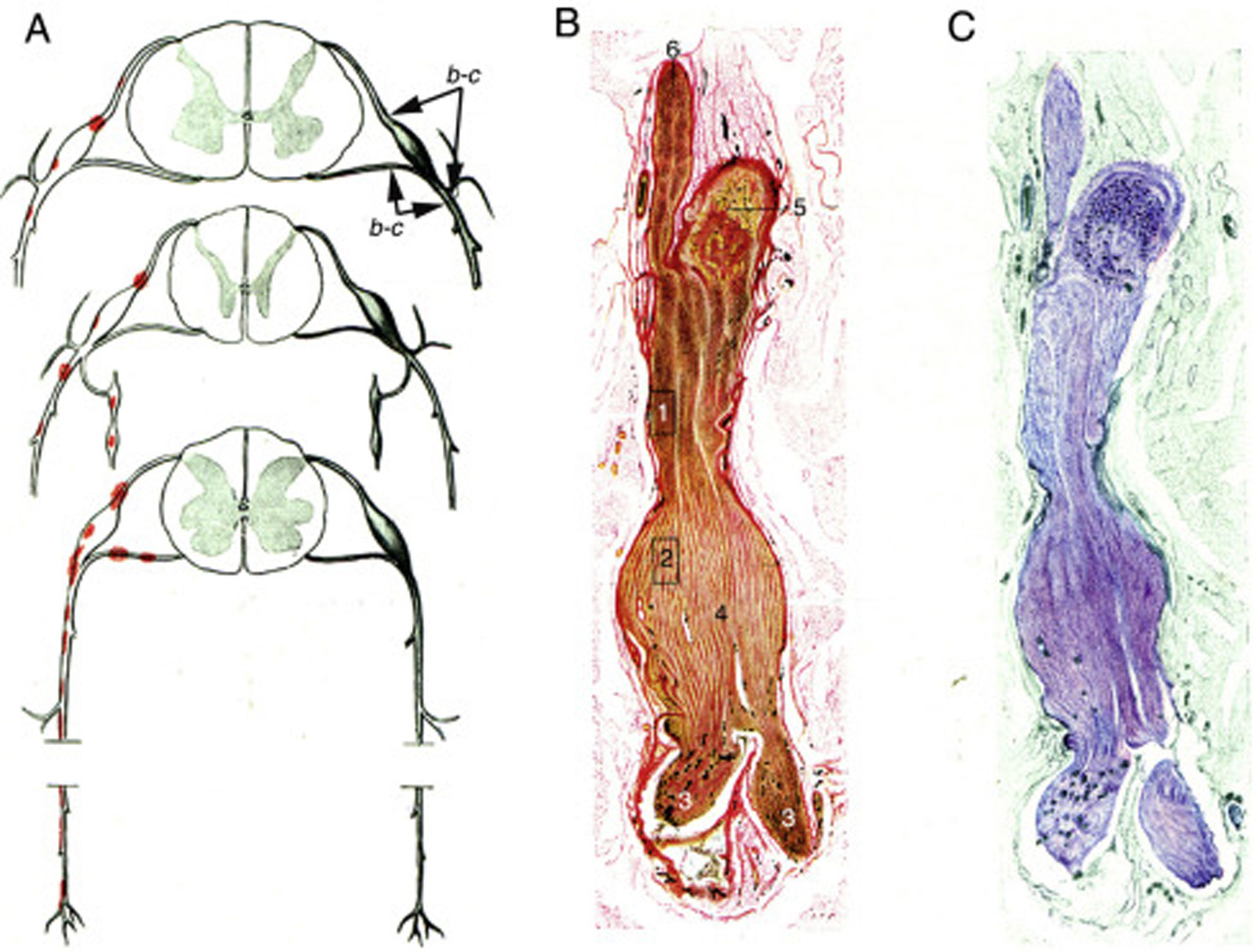
Krücke12 identificó la naturaleza inflamatoria del edema endoneural, visible a las 24h del inicio sintomático y totalmente desarrollado al tercer día, confirmando el predominio del edema inflamatorio en los troncos nerviosos proximales y particularmente en los nervios espinales (fig. 1).
Diagrama de la topografía lesional en el SGB (de arriba abajo, niveles cervical, dorsal y lumbar). Las lesiones (punteados rojos) recaen en los troncos nerviosos proximales, que incluyen raíces raquídeas ventrales y dorsales, ganglios raquídeos, ganglios simpáticos y ramos ventrales de los nervios espinales. Hemos mantenido las indicaciones b-c que el autor utilizó para señalar la localización de sus figuras 65, 66 (aquí 1B,C) y 67 (aquí no reproducida). B)Sección longitudinal de un segmento de nervio entre la raíz raquídea ventral y el nervio espinal, procedente de un paciente con SGB fallecido el día 18 de curso clínico. Hemos mantenido la numeración original. 1 y 2: áreas ilustradas por Krücke en otras figuras demostrando un extenso «edema endoneural mucoide» (entiéndase inflamatorio); 3: ramos del nervio espinal (indudablemente, ramo ventral y ramo dorsal); 4: dilatación fusiforme del nervio espinal; 5: ganglio raquídeo, y 6: raíz raquídea anterior (Van Gieson, aumento no especificado). C)Otra sección longitudinal al mismo nivel en la que se observa una coloración púrpura de la dilatación fusiforme del nervio espinal (cresil violeta).")
Reproducción con mínimas modificaciones de las figuras 65 a 67 en el trabajo de Krücke12.
A)Diagrama de la topografía lesional en el SGB (de arriba abajo, niveles cervical, dorsal y lumbar). Las lesiones (punteados rojos) recaen en los troncos nerviosos proximales, que incluyen raíces raquídeas ventrales y dorsales, ganglios raquídeos, ganglios simpáticos y ramos ventrales de los nervios espinales. Hemos mantenido las indicaciones b-c que el autor utilizó para señalar la localización de sus figuras 65, 66 (aquí 1B,C) y 67 (aquí no reproducida).
B)Sección longitudinal de un segmento de nervio entre la raíz raquídea ventral y el nervio espinal, procedente de un paciente con SGB fallecido el día 18 de curso clínico. Hemos mantenido la numeración original. 1 y 2: áreas ilustradas por Krücke en otras figuras demostrando un extenso «edema endoneural mucoide» (entiéndase inflamatorio); 3: ramos del nervio espinal (indudablemente, ramo ventral y ramo dorsal); 4: dilatación fusiforme del nervio espinal; 5: ganglio raquídeo, y 6: raíz raquídea anterior (Van Gieson, aumento no especificado).
C)Otra sección longitudinal al mismo nivel en la que se observa una coloración púrpura de la dilatación fusiforme del nervio espinal (cresil violeta).
El papel patogénico del edema endoneural en la fase precoz del síndrome fue refutado en el celebérrimo artículo de Asbury et al.13, que incluyó el estudio clínico-patológico de 19 casos fatales de SGB, cinco de ellos fallecidos entre los días 1 y 8 de curso clínico. Estos autores establecieron que el sustrato histológico del síndrome es una infiltración inflamatoria mononuclear perivenosa parcheada con desmielinización segmentaria, si bien ni macroscópica ni microscópicamente reconocieron un «estadio de edema». La relevancia fisiopatológica del edema endoneural en las neuropatías inflamatorias ha sido recientemente reconsiderada14.
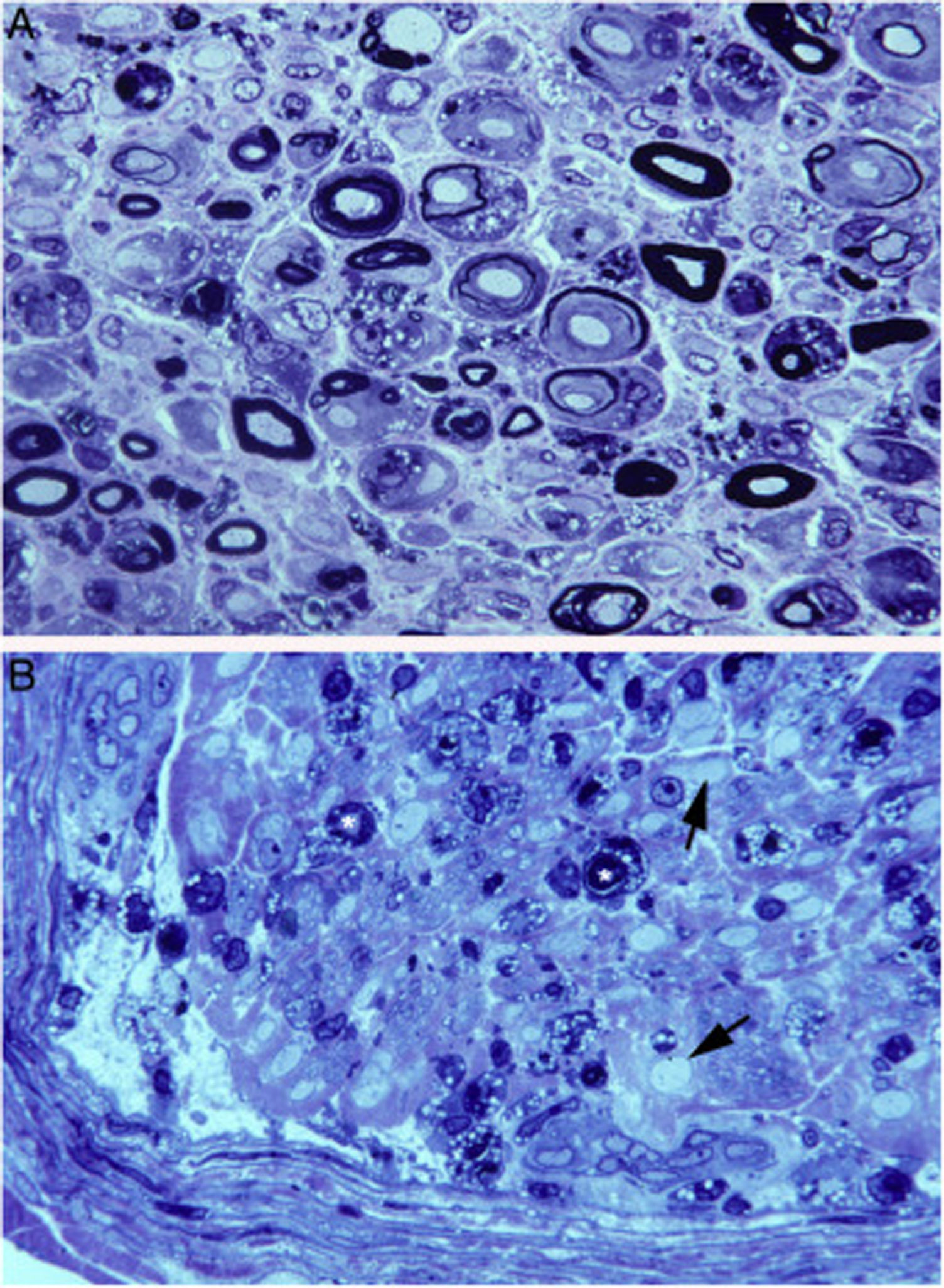
Nosotros hemos descrito un caso de AIDP fulminante que falleció al noveno día de evolución15. Las figuras 2 y 3 ilustran la patología del sistema nervioso periférico (SNP) consistente en acusado edema inflamatorio endoneural y epi-perineural con incipiente desmielinización, todo ello con predominio en los ramos ventrales de los nervios cervicales y lumbares15-17.
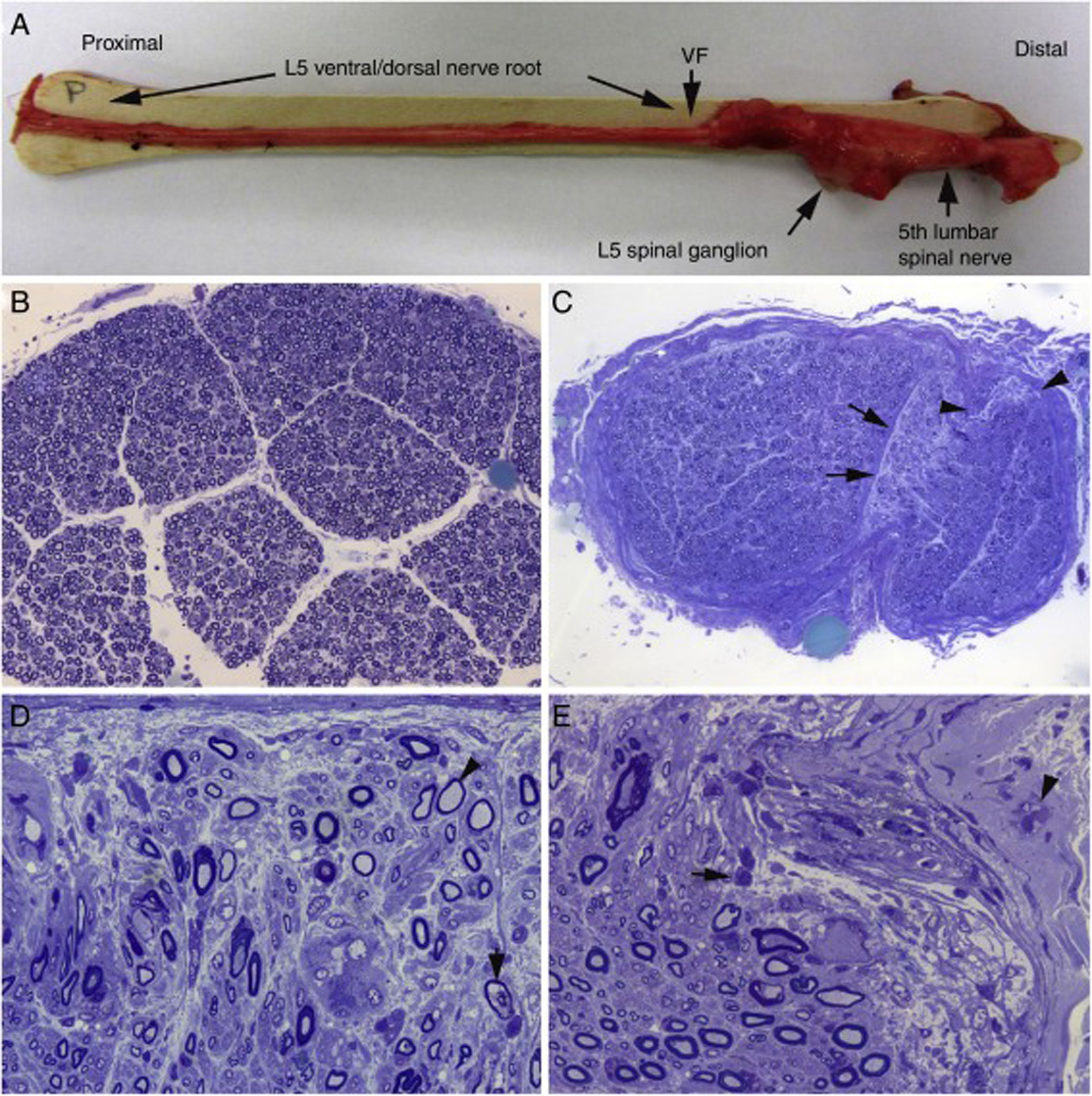
Aspecto macroscópico de L5 desde su arranque proximal hasta su entrada en el foramen vertebral (VF); la disección continúa después sin interrupción para mostrar el ensanchamiento ganglionar y el quinto nervio lumbar. Hay un engrosamiento nervioso a partir del foramen vertebral (compárese con la figura 1B, C). B)En esta sección semifina de la raíz ventral L5, a 1cm de su entrada en el foramen vertebral, se observa que la densidad de fibras mielínicas está preservada (azul de toluidina; ×100 antes de reducción). C)En esta sección semifina del ramo ventral del quinto nervio lumbar, a nivel de su emergencia foraminal, se observa edema endoneural difuso, que es más acentuado en ciertas áreas subperineurales (puntas de flecha) o en una de las regiones adyacentes al tabique peri-endoneural (flechas); el edema endoneural produce un efecto de espaciamiento de las fibras mielínicas, de manera que su densidad parece reducida en comparación con la imagen anterior (azul de toluidina; ×65 antes de reducción). D)Detalle de la zona yuxta-tabique indicada con flechas en C, que demuestra la presencia de edema inflamatorio con numerosas células mononucleares y fibras aisladas con envoltura mielínica fina para el diámetro axonal (punta de flecha) o con degeneración vacuolar de la mielina (flecha) (azul de toluidina; ×630 antes de reducción). E)Detalle del área subperineural, indicada con puntas de flecha, donde se observa un marcado edema con presencia de células inflamatorias tanto endoneurales (flecha) como epineurales (punta de flecha) (azul de toluidina; ×630 antes de reducción).")
Hallazgos histológicos en un caso fatal de AIDP15.
A)Aspecto macroscópico de L5 desde su arranque proximal hasta su entrada en el foramen vertebral (VF); la disección continúa después sin interrupción para mostrar el ensanchamiento ganglionar y el quinto nervio lumbar. Hay un engrosamiento nervioso a partir del foramen vertebral (compárese con la figura 1B, C).
B)En esta sección semifina de la raíz ventral L5, a 1cm de su entrada en el foramen vertebral, se observa que la densidad de fibras mielínicas está preservada (azul de toluidina; ×100 antes de reducción).
C)En esta sección semifina del ramo ventral del quinto nervio lumbar, a nivel de su emergencia foraminal, se observa edema endoneural difuso, que es más acentuado en ciertas áreas subperineurales (puntas de flecha) o en una de las regiones adyacentes al tabique peri-endoneural (flechas); el edema endoneural produce un efecto de espaciamiento de las fibras mielínicas, de manera que su densidad parece reducida en comparación con la imagen anterior (azul de toluidina; ×65 antes de reducción).
D)Detalle de la zona yuxta-tabique indicada con flechas en C, que demuestra la presencia de edema inflamatorio con numerosas células mononucleares y fibras aisladas con envoltura mielínica fina para el diámetro axonal (punta de flecha) o con degeneración vacuolar de la mielina (flecha) (azul de toluidina; ×630 antes de reducción).
E)Detalle del área subperineural, indicada con puntas de flecha, donde se observa un marcado edema con presencia de células inflamatorias tanto endoneurales (flecha) como epineurales (punta de flecha) (azul de toluidina; ×630 antes de reducción).
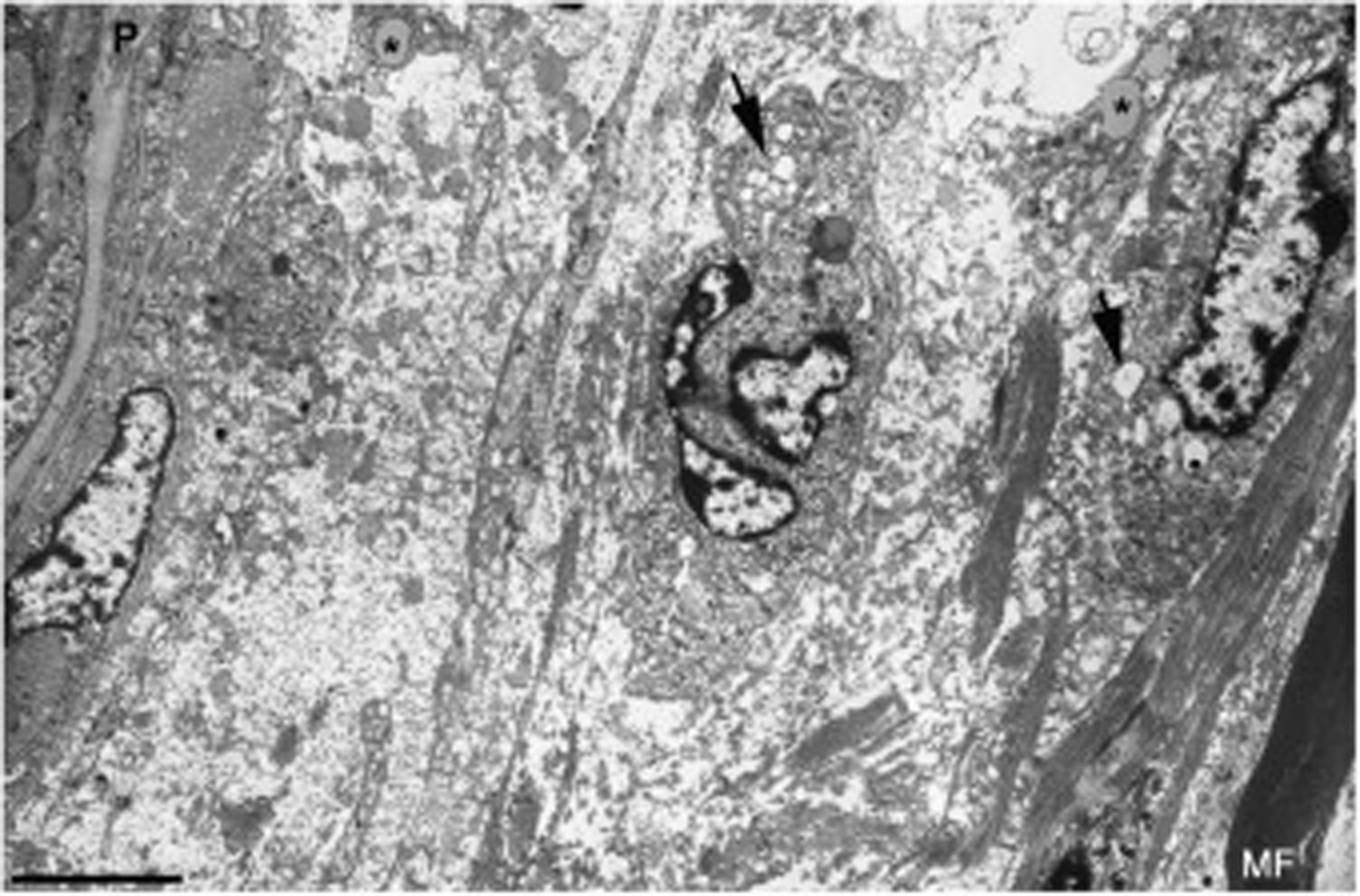
, así como numerosas vesículas de endocitosis (flechas) y lisosomas. En el área edematosa no hay fibras mielinizadas; la única observada (MF) se sitúa a 20μm del perineuro (P) (barra, 3μm).")
Imagen ultraestructual de la región superineural de la figura 2E. Hay un marcado edema endoneural con material amorfo, probablemente proteoglicano, y haces dispersos de fibrillas de colágeno. Nótese la presencia de macrófagos que contienen gotas lipídicas (asteriscos), así como numerosas vesículas de endocitosis (flechas) y lisosomas. En el área edematosa no hay fibras mielinizadas; la única observada (MF) se sitúa a 20μm del perineuro (P) (barra, 3μm).
En síntesis, todo apunta a la enorme relevancia del edema inflamatorio de los troncos nerviosos proximales en el SGB temprano.
Neuritis alérgica experimental: anotaciones seleccionadasLa neuritis alérgica experimental (NAE), inducida con diversos antígenos (eg, P0, P2, PMP22 o glucolípidos), se ha considerado el modelo animal del SGB18,19. Aquí nos ocuparemos de ciertos aspectos histológicos y neurofisiológicos en la fase inicial de la NAE inducida por el componente proteico P2 de la mielina (NAE/P2).
En la rata Lewis, la administración de 50μg de SP26 (residuo 53-78 de la proteína P2 bovina) induce una desmielinización inflamatoria pura20. Doblando la dosis de SP26 la desmielinización radicular inflamatoria se mantiene inalterada, mientras que en el nervio ciático es sustituida por degeneración axonal, de neto predominio centrofascicular21. Este cambio en la topografía lesional se relacionó con un efecto bystander a la inflamación endoneural, si bien esta es ubicua. De otro lado, la degeneración walleriana centrofascicular sugiere la sobrevenida de fenómenos isquémicos22. En un estudio secuencial de fibras nerviosas separadas en otro modelo de NAE, solo un pequeño porcentaje (del 5 al 10%) de las fibras con desmielinización inflamatoria exhibía degeneración axonal23, lo cual contradice el papel patogénico del efecto bystander.
En la NAE/P2 inducida por transferencia de linfocitos CD4+, se ha establecido que al inicio sintomático, cuarto día post-inoculación (pi), la única lesión en los troncos nerviosos es el edema inflamatorio endoneural y epineural24. Transfiriendo dosis convencionales de CD4+, la electrofisiología al quinto día es desmielinizante25. Al incrementar la dosis celular el patrón electrofisiológico evoluciona a fallo completo de la conducción nerviosa, que fue atribuida a degeneración walleriana. Ciertamente, esta interpretación es objetable, dado que en la degeneración walleriana la excitabilidad de los nervios motores solo se pierde tras 8-9días de la sección nerviosa26,27.
En la NAE/P2 se ha demostrado que la degeneración axonal histológica aparece en el cenit del edema inflamatorio endoneural y del incremento de la presión endoneural28.
Resumiendo, en la NAE/P2 puede haber una patología disociada consistente en desmielinización radicular, y degeneración axonal activa en troncos nerviosos más distantes.
Variaciones topográficas de la barrera hemato-endoneuralEl SNP está dotado de una barrera hemato-endoneural que juega un papel crucial en su homeostasis29,30. Los capilares endoneurales poseen un endotelio con uniones ocluyentes, rodeado de membrana basal y de pericitos. Experimentalmente se ha demostrado que hay diferencias topográficas en la permeabilidad vascular del SNP, de modo que los ganglios raquídeos y el endoneuro de las raíces raquídeas ventrales y dorsales hasta la unión raíz-nervio (nervio espinal) son más permeables31. Se sabe, en fin, que la barrera hemato-endoneural de los terminales nerviosos es también menos eficiente32.
Estas características de la barrera hemato-endoneural justifican la selectiva vulnerabilidad de las raíces raquídeas, ganglios raquídeos, nervios espinales y terminales nerviosos al ataque inmune, y particularmente a los mediados por anticuerpos19,33.
Síndrome de Guillain-Barré axonalClásicamente, el SGB fue considerado como ejemplo prototípico de neuropatía aguda, desmielinizante e inflamatoria, es decir, AIDP y SGB no eran sino dos designaciones para un mismo síndrome con variable grado de axonopatía secundaria13,34-36.
En 1986, Feasby et al.37 presentaron el caso de cinco pacientes con una forma grave de SGB, cuyos estudios neurofisiológicos iniciales demostraron pérdida de excitabilidad de los nervios motores y sensitivos. En tres casos (números 1, 3 y 5) tal pérdida de excitabilidad se detectó entre los días 2 y 5 de evolución. La autopsia en su caso1 puso de manifiesto una degeneración axonal primaria sin inflamación ni desmielinización, que dio paso al reconocimiento del SGB axonal. Cabe argumentar que una patología axonal pura no justifica una pérdida de excitabilidad nerviosa tan precoz.
En el estudio clínico-patológico de un caso fatal de SGB motor puro y neurofisiología axonal, nuestros estudios semifinos y ultrafinos de las raíces L5A y L5P demostraron lesiones selectivas de L5A, donde se combinaban desmielinización y degeneración axonal mediada por macrófagos (fig. 4A)38. El estudio de fibras separadas de L5A reveló signos inequívocos de desmielinización primaria22,39-41, lo cual nos llevó a proponer que el caso debía catalogarse como AIDP con degeneración axonal secundaria. Argumentamos que nuestros hallazgos en las secciones semifinas de L5A eran comparables a lo descrito por Feasby et al.37 (fig. 4A,B), y que si estos autores hubieran efectuado estudio de fibras separadas a nivel radicular quizá se habrían detectado signos de desmielinización primaria.
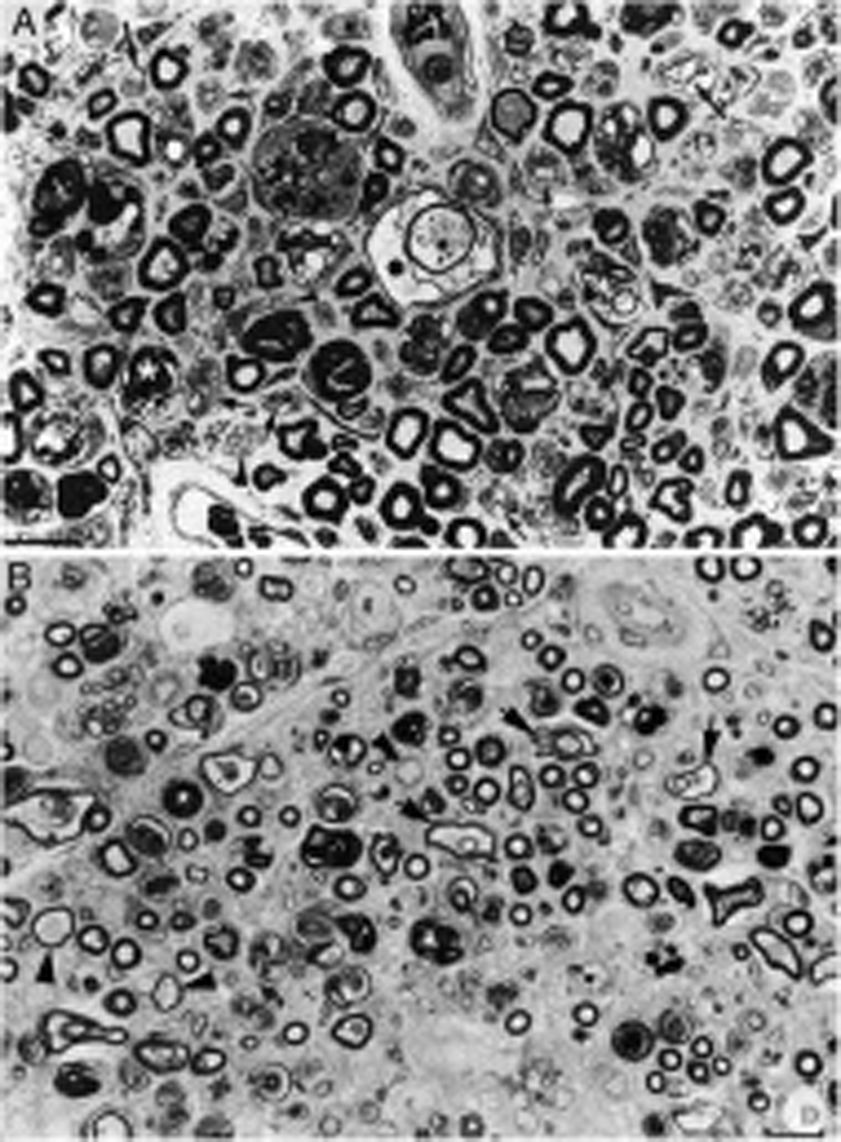
 y de la figura 2 de Feasby et al.37 (B), ambas correspondientes a secciones semifinas transversas de raíces raquídeas lumbares. A)Manteniendo los recursos gráficos originales, obsérvese la presencia de numerosos macrófagos endoneurales repletos de gotitas grasa, a veces rodeando a fibras con colapso de la mielina (asteriscos blancos) o dentro de tubos neurales totalmente desestructurados. Se observan también racimos regenerativos que contienen ya axones no mielinizados (puntas de flecha) o bien una mezcla de axones finamente mielinizados o carentes de mielina (flechas pequeñas), y ocasionales fibras más gruesas con axones desmielinizados o remielinizados (flechas grandes). En el centro de la imagen hay una fibra con disolución vesículo-vacuolar de la mielina (asterisco negro); esta lesión queda mejor documentada en la figura 6 de nuestro trabajo que muestra el elenco lesional en el estudio de fibras separadas38 (azul de toulidina; barra=19μm). B)En la leyenda original de esta figura, sin recursos gráficos, los autores escriben lo siguiente: «sección transversa que muestra acusada degeneración axonal». Nuestra lectura coincide con la de los autores, porque efectivamente hay numerosos axones con colapso de la mielina (asteriscos blancos), lo cual es indicativo de degeneración axonal aguda22,39. Obsérvese además la presencia de fibras con mielina excesivamente fina para el diámetro axonal (puntas de flechas) y fibras que exhiben degeneración vacuolar de la mielina (flechas negras), todo ello sugestivo de des-remielinización, y de racimos regenerativos. A nuestro entender, en el intersticio endoneural se identifican células inflamatorias mononucleares, cuya presencia se hubiera valorado mejor mediante técnicas inmunocitoquímicas22,39. Aunque la disolución vesicular de la mielina puede ser un artefacto post mórtem40, en nuestro material se observó solo en L5A (lesionada) y no en L5P (indemne), un hecho que apoya su valor morfológico como signo de desmielinización39. Además, la degeneración vesicular de la mielina en la AIDP es el signo más precoz, presente incluso cuando todavía no hay macrófagos en el tubo neural, lo cual sugiere que es la expresión de la formación del complejo de ataque a membrana por activación de la cascada del complemento41 (barra=20μm). Reproducida con el permiso editorial de Brain.")
Composición de la figura 3 de Berciano et al.38 (A) y de la figura 2 de Feasby et al.37 (B), ambas correspondientes a secciones semifinas transversas de raíces raquídeas lumbares.
A)Manteniendo los recursos gráficos originales, obsérvese la presencia de numerosos macrófagos endoneurales repletos de gotitas grasa, a veces rodeando a fibras con colapso de la mielina (asteriscos blancos) o dentro de tubos neurales totalmente desestructurados. Se observan también racimos regenerativos que contienen ya axones no mielinizados (puntas de flecha) o bien una mezcla de axones finamente mielinizados o carentes de mielina (flechas pequeñas), y ocasionales fibras más gruesas con axones desmielinizados o remielinizados (flechas grandes). En el centro de la imagen hay una fibra con disolución vesículo-vacuolar de la mielina (asterisco negro); esta lesión queda mejor documentada en la figura 6 de nuestro trabajo que muestra el elenco lesional en el estudio de fibras separadas38 (azul de toulidina; barra=19μm).
B)En la leyenda original de esta figura, sin recursos gráficos, los autores escriben lo siguiente: «sección transversa que muestra acusada degeneración axonal». Nuestra lectura coincide con la de los autores, porque efectivamente hay numerosos axones con colapso de la mielina (asteriscos blancos), lo cual es indicativo de degeneración axonal aguda22,39. Obsérvese además la presencia de fibras con mielina excesivamente fina para el diámetro axonal (puntas de flechas) y fibras que exhiben degeneración vacuolar de la mielina (flechas negras), todo ello sugestivo de des-remielinización, y de racimos regenerativos. A nuestro entender, en el intersticio endoneural se identifican células inflamatorias mononucleares, cuya presencia se hubiera valorado mejor mediante técnicas inmunocitoquímicas22,39. Aunque la disolución vesicular de la mielina puede ser un artefacto post mórtem40, en nuestro material se observó solo en L5A (lesionada) y no en L5P (indemne), un hecho que apoya su valor morfológico como signo de desmielinización39. Además, la degeneración vesicular de la mielina en la AIDP es el signo más precoz, presente incluso cuando todavía no hay macrófagos en el tubo neural, lo cual sugiere que es la expresión de la formación del complejo de ataque a membrana por activación de la cascada del complemento41 (barra=20μm).
Reproducida con el permiso editorial de Brain.
Nuestro trabajo38 se siguió de una viva controversia en tres artículos consecutivos en la sección «Issues&Opinions» de Muscle and Nerve (junio de 1994); en uno de ellos se planteó la dificultad para distinguir neurofisiológicamente una axonopatía primaria de otra secundaria a desmielinización inflamatoria42, mientras que en los dos restantes los autores se enrocaron en torno a la individualidad nosológica del SGB axonal43,44 y de la AMAN45. En otro caso posterior con SGB motor fulminante y neurofisiología axonal, el examen autópsico demostró desmielinización de las raíces y nervios espinales, e indemnidad de los nervios surales y medianos46,47.
Resumiendo, hay un indudable solapamiento entre el SGB axonal y la AIDP con degeneración axonal secundaria, cuya distinción a veces solo es posible tras estudios autópsicos detallados.
AMAN y su conexión con las nodo-paranodopatíasEn 1991, McKhann et al.48 describieron un síndrome de parálisis flácida aguda (PFA) estival en áreas rurales del norte de China. Aunque considerado un cuadro motor puro, muchos pacientes tenían dolor y signos meníngeos. Los resultados de los primeros 10casos fatales de PFA se encasillaron en tres grandes patrones: a)cinco casos con degeneración walleriana de los raíces raquídeas anteriores y fibras motoras de los nervios periféricos; b)tres casos con desmielinización inflamatoria, y c)dos casos sin lesiones patogénicas49. Para la PFA axonal se introdujo la designación de AMAN. Con enorme precisión, los autores describen que la lesión en AMAN se inicia en la porción proximal o intermedia de la raíz ventral, incrementando hasta su punto de emergencia a través de la dura, donde la degeneración alcanza al 80% de las fibras. En el 90% de los sueros de sus pacientes, recolectados entre los días 1 y 10 de curso clínico, se detectaron anticuerpos frente a Campylobacter jejuni, una asociación patogénica ya establecida una década antes50.
En una serie de trabajos posteriores, el Grupo del Profesor Jack Griffin completó la descripción histológica e inmunopatológica de la PFA con 12 estudios necrópsicos adicionales41,51-55. Se distinguieron de nuevo tres patrones neuropatológicos básicos: axonal en 6 (3 AMAN y 3 AMSAN), AIDP en 3, y mínima patología en 3. Se propuso que la AMAN es un nuevo síndrome mediado por un ataque inmune contra el axolema nodal de las fibras motoras mediado por anticuerpos antigangliósido y activación del complemento54. En las dos últimas décadas la investigación sobre la relación patogénica entre gangliósido y SGB ha sido una cuestión destacada e incesante (1006 Search results en PubMed, acceso 27 de abril de 2018), abordada en recientes revisiones3,9,56-58.
Los avances en el conocimiento de la arquitectura molecular de la fibra nerviosa y el descubrimiento de dianas antigénicas nodales y paranodales59 han dado paso a una nueva fisiopatología de las neuropatías autoinmunes, alumbrando el concepto de nodo-paranodopatías60-64. En la AMAN el ataque inmune frente a epitopos nodales tiene la siguientes consecuencias: a)alteración de los canales Nav nodales, que puede llevar aparejado un potencial fallo reversible a la conducción sin fenómenos desmielinizantes acompañantes, cuya constatación obliga a estudios neurofisiológicos consecutivos; b)invasión macrofágica de la región nodal del tubo neural, potencial causa de disfunción de los tabiques axo-gliales paranodales adyacentes con disrupción de la mielina paranodal, y c)cuando el complejo de ataque a la membrana alcanza un nivel crítico, entrada masiva de Ca++ en el axoplasma con degeneración axonal activa.
El concepto de nodo-paranodopatía abre perspectivas patogénicas inéditas, que son de suma importancia para la correcta interpretación de las alteraciones neurofisiológicas en el SGB.
La forma motora «pura» del síndrome de Guillain-Barré: ¿solo motora y siempre axonal?Desde la descripción original65, se sabe que el cuadro clínico del SGB suele ser preferentemente motor.
En 27 (18%) de sus 147 pacientes de SGB, Visser et al.66 observaron una semiología casi exclusivamente motora y neurofisiología axonal. Comparativamente con los restantes pacientes de su serie, este subgrupo motor tenía además ciertas características específicas, de modo que este fue considerado como una entidad afín a la AMAN, aunque sin su carácter epidémico estival. A partir de este influyente trabajo, SGB motor puro, SGB axonal y AMAN fueron a menudo designaciones usadas indistintamente.
En la serie de Hadden et al.67, que incluyó 369 pacientes de SGB, hubo 53 (16%) con SGB motor «puro» tipificados del siguiente modo: a)27 desmielinizantes (10% de los catalogados desmielinizantes); b)5 axonales (50% de los catalogados axonales), y c)21 con neurofisiología inexcitable, equívoca o normal.
Así pues, aunque el subtipo motor «puro» ocurra con mayor frecuencia entre pacientes con SGB axonal, este puede darse en pacientes con AIDP, incluyendo casos con comprobación necrópsica (vide supra)38,46,47.
Determinismo patogénico del epi-perineuro en el inicio lesional del síndrome de Guillain-BarréHemos visto que, al incrementar la dosis de inmunógeno, en la NAE/P2 hay una discordancia lesional entre raíces raquídeas y nervio ciático20,21. También hemos visto el predominio lesional en las raíces raquídeas y nervios espinales en estudios necrópsicos de pacientes con SGB fallecidos en los primeros días de curso clínico. Llegados a este punto, cabe plantear la siguiente interrogante: ¿cómo explicar que una patología desmielinizante proximal sea sustituida por una axonal en troncos nerviosos más distantes? Nuestros estudios clínico-patológicos en el SGB han tratado de resolver este enigma.
En un paciente de 67 años con SGB fulminante los estudios neurofisiológicos consecutivos (días 3, 10 y 17 del inicio) demostraron una pérdida de la excitabilidad nerviosa68. El estudio histológico demostró las lesiones ilustradas en la figura 5, básicamente consistentes en desmielinización masiva de las raíces raquídeas, y en una patología mixta, axonal y demielinizante, de troncos nerviosos más distantes. Así pues, estos hallazgos reproducían fidedignamente la patología disociada entre raíces raquídeas y troncos nerviosos periféricos descrita en la NAE/P220,21. Partiendo de estos datos y de la anatomía microscópica de la médula espinal con sus raíces raquídeas y nervios espinales (fig. 6)16,69, nos planteamos la necesidad de llevar a cabo estudios histológicos consecutivos de raíces, ramos ventrales de los nervios espinales, y troncos nerviosos más distantes. Supusimos que en las raíces raquídeas, dotadas de una laxa envuelta aracnoidea, la desmielinización inflamatoria radicular inicial puede ser acomodada sin que haya un incremento de la presión endoneural. A partir del ángulo subaracnoideo, la duramadre pasa a ser epineuro y la aracnoides se transforma en perineuro, que dan envoltura a los troncos nerviosos periféricos hasta sus terminales motores y sensitivos. Siendo la compliancia del epi-perineuro menor que la de la aracnoides, supusimos también que una inflamación endoneural crítica, en fases precoces del SGB, podría ser acomodada solamente a expensas de un incremento de la presión endoneural, potencial causa de un compromiso del flujo vascular transperineural, y responsable a la postre de la isquemia endoneural con el consiguiente fallo en la conducción nerviosa (ver fig. 6).
Sección semifina de L5A en la que se observa una desmielinización masiva con numerosos macrófagos repletos de gotitas de grasa. B)Sección semifina del nervio crural en la que se observan numerosas fibras con colapso de la mielina (asteriscos), lo cual es indicativo de degeneración axonal activa, y axones desmielinizados (flechas). Nótese de nuevo la presencia de macrófagos repletos de gotitas de grasa, a menudo rodeando fibras con mielina colapsada (azul de toulidina, ×630 antes de reducción).")
Patología en el SGB fulminante con inexcitabilidad precoz de los nervios69.
A)Sección semifina de L5A en la que se observa una desmielinización masiva con numerosos macrófagos repletos de gotitas de grasa.
B)Sección semifina del nervio crural en la que se observan numerosas fibras con colapso de la mielina (asteriscos), lo cual es indicativo de degeneración axonal activa, y axones desmielinizados (flechas). Nótese de nuevo la presencia de macrófagos repletos de gotitas de grasa, a menudo rodeando fibras con mielina colapsada (azul de toulidina, ×630 antes de reducción).
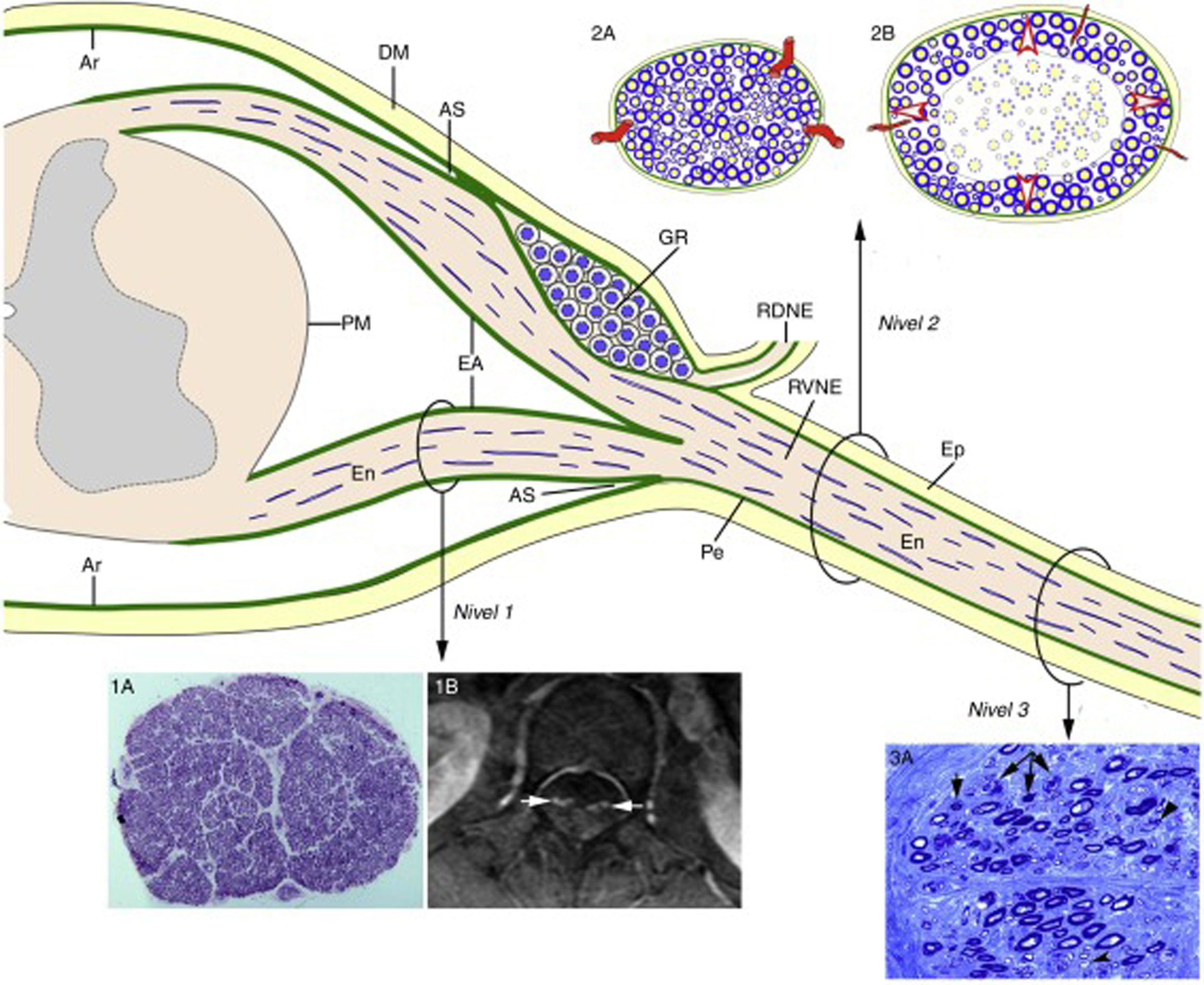
, el epineuro (Ep) está en continuidad con la duramadre (DM). El endoneuro (En) de los troncos nerviosos periféricos se prolonga por las raíces hasta su unión con la médula espinal. En el AS, gran parte del perineuro (Pe) se incorpora a la aracnoides subdural (Ar), pero unas pocas capas forman la envoltura aracnoidea (EA) de las raíces espinales. La Ar radicular se continúa con la piamadre (PM) en su emergencia de la médula espinal. Inmediatamente después del ganglio espinal raquídeo (GR), en el AS, las raíces ventrales y dorsales se unen para formar el nervio espinal, que emerge por el agujero vertebral dividiéndose en un ramo dorsal (RDNE) y otro ventral (RVNE). Consecuentemente, las raíces raquídeas intratecales poseen una envoltura laxa derivada de la aracnoides, mientras que la de los nervios espinales y de los troncos nerviosos más distantes consta de epi-perineuro que es relativamente inelástica. Las lesiones inflamatorias próximo-distales en la fase precoz del SGB se ilustran del siguiente modo: raíz ventral lumbar (Nivel 1), nervio espinal (Nivel 2) y nervio ciático (Nivel 3). En el Nivel 1, esta sección semifina transversa completa de la raíz L5, en un caso de SGB fatal, demuestra preservación de la densidad de fibras mielinizadas (1A), aunque las lesiones inflamatorias observables a mayor aumento (no mostradas) pueden justificar el incremento del área transversa, y su engrosamiento y realce tras la administración de contraste en el examen de resonancia magnética (1B, flechas). Los dos dibujos del Nivel 2 ilustran los siguientes hechos: a)anatomía normal de un nervio espinal, usualmente monofascicular, con sus vasos transperineurales y su envoltura epi-perineural (2A), que justifican su aspecto ultrasonográfico normal caracterizado por una estructura redondeada u ovalada hipoecoica con un anillo periférico hipercoico (ver texto más adelante), y b)en la fase precoz del SGB el edema inflamatorio endoneural puede provocar una elevación crítica de la presión endoneural en los nervios espinales, que tensa el epi-perineuro más allá de su compliancia constriñendo los vasos transperineurales, lo cual conduce a la isquemia endoneural, aquí centrofascicular (2B). El Nivel 3 corresponde a una sección semifina de nervio ciático en un caso de AIDP fatal, donde se observan varias fibras con degeneración axonal (colapso de la mielina, flechas), que aquí es fenómeno secundario a lesiones desmielinizantes inflamatorias más proximales; nótese también la presencia de fibras remielinizadas (puntas de flecha) y de macrófagos repletos de grasa. Sin conocer la existencia de una patología desmielinizante proximal, hubiera sido muy difícil interpretar fidedignamente el papel patogénico de la florida patología axonal observada en esta imagen. Diagrama inspirado por la figura 2-6 del trabajo de Berthold y colaboradores68.")
Diagrama de la anatomía microscópica de la médula espinal, raíces raquídeas y nervios espinales según Berciano et al.16. A partir del ángulo subaracnoideo (AS), el epineuro (Ep) está en continuidad con la duramadre (DM). El endoneuro (En) de los troncos nerviosos periféricos se prolonga por las raíces hasta su unión con la médula espinal. En el AS, gran parte del perineuro (Pe) se incorpora a la aracnoides subdural (Ar), pero unas pocas capas forman la envoltura aracnoidea (EA) de las raíces espinales. La Ar radicular se continúa con la piamadre (PM) en su emergencia de la médula espinal. Inmediatamente después del ganglio espinal raquídeo (GR), en el AS, las raíces ventrales y dorsales se unen para formar el nervio espinal, que emerge por el agujero vertebral dividiéndose en un ramo dorsal (RDNE) y otro ventral (RVNE). Consecuentemente, las raíces raquídeas intratecales poseen una envoltura laxa derivada de la aracnoides, mientras que la de los nervios espinales y de los troncos nerviosos más distantes consta de epi-perineuro que es relativamente inelástica. Las lesiones inflamatorias próximo-distales en la fase precoz del SGB se ilustran del siguiente modo: raíz ventral lumbar (Nivel 1), nervio espinal (Nivel 2) y nervio ciático (Nivel 3). En el Nivel 1, esta sección semifina transversa completa de la raíz L5, en un caso de SGB fatal, demuestra preservación de la densidad de fibras mielinizadas (1A), aunque las lesiones inflamatorias observables a mayor aumento (no mostradas) pueden justificar el incremento del área transversa, y su engrosamiento y realce tras la administración de contraste en el examen de resonancia magnética (1B, flechas). Los dos dibujos del Nivel 2 ilustran los siguientes hechos: a)anatomía normal de un nervio espinal, usualmente monofascicular, con sus vasos transperineurales y su envoltura epi-perineural (2A), que justifican su aspecto ultrasonográfico normal caracterizado por una estructura redondeada u ovalada hipoecoica con un anillo periférico hipercoico (ver texto más adelante), y b)en la fase precoz del SGB el edema inflamatorio endoneural puede provocar una elevación crítica de la presión endoneural en los nervios espinales, que tensa el epi-perineuro más allá de su compliancia constriñendo los vasos transperineurales, lo cual conduce a la isquemia endoneural, aquí centrofascicular (2B). El Nivel 3 corresponde a una sección semifina de nervio ciático en un caso de AIDP fatal, donde se observan varias fibras con degeneración axonal (colapso de la mielina, flechas), que aquí es fenómeno secundario a lesiones desmielinizantes inflamatorias más proximales; nótese también la presencia de fibras remielinizadas (puntas de flecha) y de macrófagos repletos de grasa. Sin conocer la existencia de una patología desmielinizante proximal, hubiera sido muy difícil interpretar fidedignamente el papel patogénico de la florida patología axonal observada en esta imagen.
Diagrama inspirado por la figura 2-6 del trabajo de Berthold y colaboradores68.
En tres estudios clínico-patológicos posteriores abordamos si la patología desmielinizante inflamatoria radicular se modifica en el tránsito de raíz raquídea a nervio espinal15,70,71. Se observó un drástico cambio del patrón lesional de raíz L5A a ramo ventral del quinto nervio espinal (ver figs. 2 y 3). Parecidos cambios secuenciales se observaron en los otros dos pacientes de AIDP, que inicialmente exhibieron un patrón neurofisiológico axonal. La figura 7 ilustra además que en los troncos nerviosos dotados de epi-perineuro puede haber áreas en cuña o centrofasciculares con una acusada reducción de fibras mielínicas gruesas, lo cual sugiere que han sobrevenido fenómenos isquémicos endoneurales22.
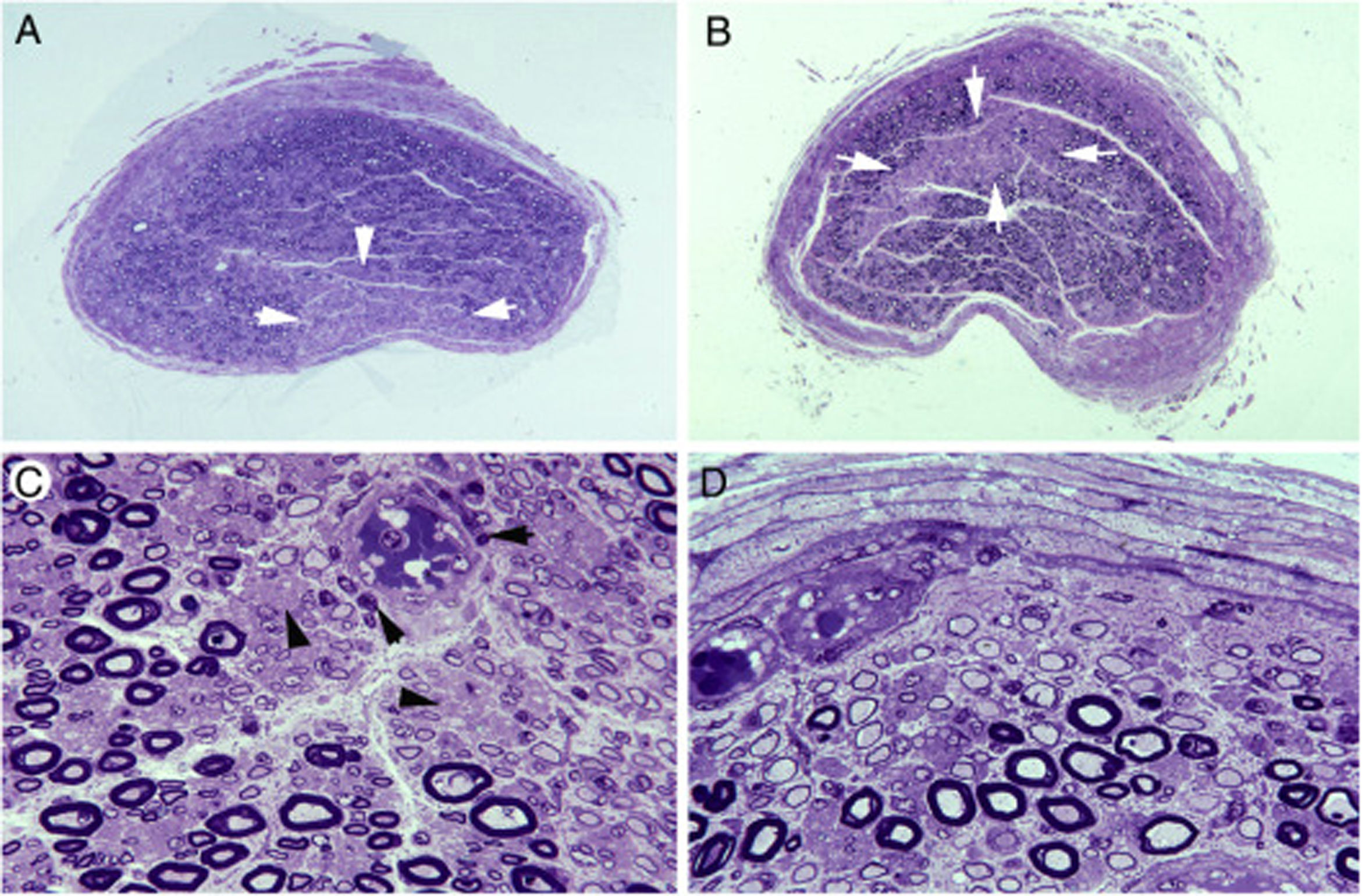
Sección transversa semifina del tercer nervio lumbar en la que se observa un área en cuña (flechas) con acusada pérdida de fibras mielínicas (azul de toluidina; ×62 antes de reducción). B)Sección transversa semifina del tronco lumbosacro en la que hay un área centrofascicular (flechas) con pérdida de fibras mielínicas (azul de toluidina; ×62 antes de reducción). Tanto en A como en B, nótese una reducción difusa de la densidad de fibras mielínicas. C)Detalle de la región centrofascicular del tronco lumbosacro a más aumento, donde se observa una acusada pérdida de fibras mielínicas gruesas, pequeños axones finamente mielinizados, axonales amielínicos preservados (puntas de flecha) y numerosas células inflamatorias mononucleares, algunas con distribución perivascular (flechas) (azul de toluidina; ×375 antes de reducción). D)En esta región subperineural del tronco lumbosacro, obsérvese numerosas fibras des-remielinizadas y numerosas células mononucleares; tan extensa des-remielinización justifica, en buena medida, la aparente pérdida de fibras mielinizadas en A y B (azul de toluidina; ×475 antes de reducción).")
Lesiones isquémicas en troncos nerviosos proximales en la AIDP70.A)Sección transversa semifina del tercer nervio lumbar en la que se observa un área en cuña (flechas) con acusada pérdida de fibras mielínicas (azul de toluidina; ×62 antes de reducción).
B)Sección transversa semifina del tronco lumbosacro en la que hay un área centrofascicular (flechas) con pérdida de fibras mielínicas (azul de toluidina; ×62 antes de reducción). Tanto en A como en B, nótese una reducción difusa de la densidad de fibras mielínicas.
C)Detalle de la región centrofascicular del tronco lumbosacro a más aumento, donde se observa una acusada pérdida de fibras mielínicas gruesas, pequeños axones finamente mielinizados, axonales amielínicos preservados (puntas de flecha) y numerosas células inflamatorias mononucleares, algunas con distribución perivascular (flechas) (azul de toluidina; ×375 antes de reducción).
D)En esta región subperineural del tronco lumbosacro, obsérvese numerosas fibras des-remielinizadas y numerosas células mononucleares; tan extensa des-remielinización justifica, en buena medida, la aparente pérdida de fibras mielinizadas en A y B (azul de toluidina; ×475 antes de reducción).
Resumiendo, el tránsito de raíz raquídea a nervio espinal implica un cambio drástico de la patología precoz en la AIDP con aparición de áreas de isquemia endoneural, lo cual indica el determinismo patogénico del edema inflamatorio en troncos nerviosos dotados de epi-perineuro.
Neurofisiología en la fase precoz del síndrome de Guillain-BarréLos estudios de conducción nerviosa son la prueba diagnóstica esencial no solo para establecer que el síndrome paralítico es neuropático, sino para fijar el subtipo de SGB3-6,56. En los primeros días de curso clínico los estudios neurofisiológicos a menudo revelan hallazgos no indicativos de un patrón desmielinizante o axonal, tales como alteración de las ondasF o de la respuestaH2,72-78. En los cuatro primeros días de curso clínico y en función de los criterios electrodiagnósticos utilizados, la sensibilidad de los estudios de conducción nerviosa para la AIDP pueden variar entre el 19 y el 63%75.
Mediante una técnica de estimulación de las raíces lumbares y en 15 pacientes de SGB temprano, Kurt Incesu et al.79 observaron que las respuestas M eran siempre de reducida amplitud, mientras que los estudios neurofisiológicos convencionales eran normales o no diagnósticos en 6 (40%) de ellos. Desde otra perspectiva, en 12 pacientes con SGB temprano Temuçin y Nurlu80 describieron una prolongación significativa del tiempo de conducción de la raíz motora en el 83% de los casos. Se ha propuesto que el tiempo de conducción de la cola de caballo está frecuentemente alterado en la AIDP, pero preservado en la AMAN81. Este resultado está en aparente contradicción con lo descrito por Sevy et al.82, quienes, usando la técnica de la triple estimulación en seis pacientes de AMAN temprana cuyos exámenes neurofisiológicos convencionales no fueron diagnósticos, registraron un bloqueo proximal a la conducción constante, que se sitúa en la emergencia foraminal del ramo ventral del nervio espinal. En 13 pacientes con SGB temprano, la estimulación desde punto de Erb con registro en mediano y cubital determinó alteraciones de la latencia y amplitud de los PAMC en 10 (77%)83.
Los resultados de todos estos estudios neurofisiológicos indican la enorme relevancia patogénica del bloqueo motor proximal en los estadios iniciales del SGB, lo cual está en perfecta armonía con la topografía lesional, tanto en la AIDP como en la AMAN.
Pruebas de imagen en la fase precoz del síndrome de Guillain-BarréLa resonancia magnética (RM) medular y la ultrasonografía (US) de troncos nerviosos han sido las dos pruebas por imagen de elección en la fase aguda del SGB. Las analizaremos por separado.
Resonancia magnética espinalEn no pocos casos el examen de RM espinal se ha indicado al ingreso del paciente con el fin de establecer un diagnóstico diferencial entre parálisis ascendente por SGB o por mielopatía aguda84. En varias series de pacientes con SGB se ha detectado casi constantemente realce radicular tras la administración de contraste38,85-89 (para revisión, ver Berciano et al.16). Tiene interés señalar que este realce puede circunscribirse a las raíces ventrales en la forma motora pura89 y en la forma paraparética y axonal90-92.
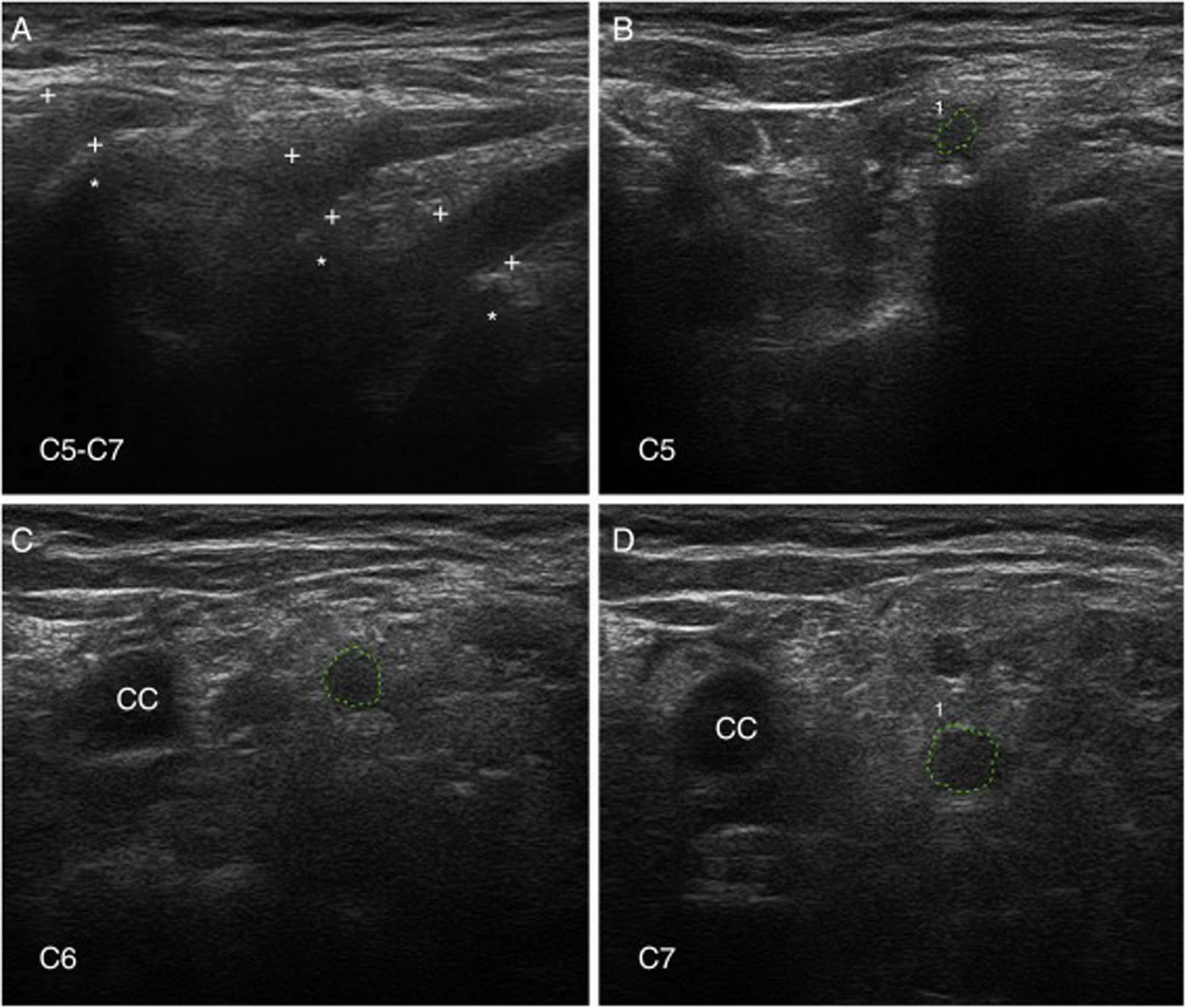
Ultrasonografía de nervioLa US de nervio es actualmente una técnica diagnóstica de rutina para el estudio de las enfermedades del SNP93. En nuestra serie de seis pacientes de SGB temprano, con cuatro casos catalogados como AIDP y los dos restantes como AMSAN, las principales alteraciones US ocurrieron en los ramos ventrales de los nervios cervicales, consistentes en incremento del área seccional transversa, borramiento de su anillo hiperecoico epineural, o ambos (fig. 8)15,94. En solo un 8,8% de los sonogramas de troncos nerviosos periféricos se observaron alteraciones inflamatorias, que afectaban sobre todo al nervio mediano.
 de nervios cervicales en la AIDP (mismo caso de la figuras 2 y 3); para hallazgos histológicos en el sexto nervio cervical, ver figura 1 y S1 en la referencia de Gallardo et al.15. A)Sonograma sagital en el que se observa un borramiento de la envoltura epineural de los tres nervios escaneados (calibradores señalados con cruces). Los asteriscos indican las apófisis vertebrales transversas. B-D)Sonogramas de los ejes cortos de los ramos ventrales de los nervios C5-C7, que han sido delimitados por trazos interrumpidos verdes. Las áreas seccionales transversas estaban significativamente incrementadas. Nótese la ausencia del anillo hiperecoico epineural, lo cual puede contrastarse con las imágenes normales en el trabajo de Haun et al.94. El edema inflamatorio endoneural (ver figs. 2C-D y 3) justifica el incremento del área seccional transversa, mientras que el componente inflamatorio epi-perineural (ver fig. 2E) responde del borramiento del anillo hiperecoico epineural.")
Ultrasonografía (US) de nervios cervicales en la AIDP (mismo caso de la figuras 2 y 3); para hallazgos histológicos en el sexto nervio cervical, ver figura 1 y S1 en la referencia de Gallardo et al.15.
A)Sonograma sagital en el que se observa un borramiento de la envoltura epineural de los tres nervios escaneados (calibradores señalados con cruces). Los asteriscos indican las apófisis vertebrales transversas.
B-D)Sonogramas de los ejes cortos de los ramos ventrales de los nervios C5-C7, que han sido delimitados por trazos interrumpidos verdes. Las áreas seccionales transversas estaban significativamente incrementadas. Nótese la ausencia del anillo hiperecoico epineural, lo cual puede contrastarse con las imágenes normales en el trabajo de Haun et al.94. El edema inflamatorio endoneural (ver figs. 2C-D y 3) justifica el incremento del área seccional transversa, mientras que el componente inflamatorio epi-perineural (ver fig. 2E) responde del borramiento del anillo hiperecoico epineural.
A diferencia de nuestros hallazgos, otros autores han observado un incremento de las áreas seccionales de los troncos nerviosos periféricos en fases precoces del SGB, si bien solo el edema regresivo de los nervios C5-C6 parece correlacionarse con la mejoría clínica95-97. Dado que la US de nervio es una técnica dependiente de la pericia del explorador, son necesarios más estudios para determinar con mayor precisión su valor dentro del elenco de pruebas diagnósticas en el SGB98,99.
Consideraciones diagnósticasLa patología predominante de los nervios espinales en la fase muy precoz (≤4días) de la AIDP justifica que, pese a que exista una grave parálisis, los estudios de conducción nerviosa convencionales puedan ser normales o no diagnósticos. La evolución posterior abre dos grandes rutas16,90: a)que se produzca una isquemia endoneural masiva en los nervios espinales que comporte la correspondiente axonopatía, en cuyo caso el patrón neurofisiológico inicial dará paso a otro axonal, y b)que sobrevengan las esperables lesiones desmielinizantes a lo largo de los troncos nerviosos, lo cual abocará a un patrón neurofisiológico desmielinizante.
Por lo que respecta a la AMAN temprana, el edema inflamatorio en los troncos nerviosos proximales puede tener un impacto comparable al señalado para la AIDP. Aquí, si el síndrome asocia anticuerpos antigangliósido, estos pueden jugar un papel patogénico ocasionando bloqueo reversible de la conducción no desmielinizante, a nivel de troncos nerviosos proximales, intermedios o distales. En AMAN/AMSAN, la evolución de disfunción axonal a degeneración walleriana dependerá tanto del impacto del edema inflamatorio inicial como del grado de disrupción del axolema nodal/paranodal inducido por los anticuerpos antigangliósido60-62. Sea como fuere, los estudios neurofisiológicos consecutivos son imprescindibles para asesorar el bloqueo reversible no desmielinizante de la conducción y el patrón axonal evolutivo100.
Consideraciones terapéuticasEl tratamiento medicamentoso específico del SGB descansa en el uso de IGIV o de plasmaféresis5,6,56. De forma sistemática se desaconseja el tratamiento con corticoides5,6,56. Hay, sin embargo, dos situaciones que merecen consideración aparte.
La primera es la del paciente con una forma fulminante de SGB y pérdida temprana de excitabilidad de los troncos nerviosos69. El papel patogénico del edema endoneural en este supuesto es tan relevante como el que subyace en cualquier hipertensión intracraneal descompensada16,101. En estas circunstancias, nos parece totalmente justificada la administración de pulsos intravenosos de metilprednisolona o de dexametasona. Por extensión, cabe plantear el futuro uso de otras terapias anti-edema.
La segunda situación se relaciona con la semiología del dolor. Tanto en la AIDP como en la AMAN, el dolor de tronco nervioso102 es una manifestación frecuente que en un tercio de los casos precede la aparición de parálisis103-105. El dolor se ha asociado con la inflamación de las raíces posteriores, lo cual es aplicable para la AIDP pero no así para la AMAN, donde, por definición, tales raíces están indemnes. Como hemos visto, en la AMAN sistemáticamente hay una patología de las raíces ventrales que se prolonga hasta los ramos ventrales de los nervios espinales. En contigüidad con estos ramos, se sitúan los ramos dorsales (ver fig. 6). El edema inflamatorio inicial de los ramos ventrales de los nervios espinales puede alcanzar a sus congéneres dorsales por un doble mecanismo16: a)al ser una estructura adyacente, por mera continuidad del proceso inflamatorio, y b)por incremento de la presión endoneural en el nervio espinal con isquemia endoneural, que inevitablemente afectará a las fibras destinadas a los ramos dorsales. Esta base inflamatoria justifica el uso de corticoides para el tratamiento de dolor rebelde en el SGB; hay 13 trabajos bien documentados de pacientes con acusado y rebelde dolor de espalda y buena respuesta a los corticoides16.
ConclusiónEn la fase precoz del SGB hay edema inflamatorio de los troncos nerviosos proximales, que puede condicionar fallo de la conducción nerviosa y degeneración axonal activa.
FinanciaciónEste trabajo no recibió ninguna subvención específica de organismos públicos ni entidades privadas.
Conflictos de interesesEl autor declara que no existe conflicto de intereses.
Este trabajo resume tres décadas de estudios dedicados al SGB, en las que he tenido la inestimable ayuda de mis colegas, particularmente de la Dra. M.ª José Sedano, del Servicio de Neurología en el Hospital Universitario Marqués de Valdecilla (HUMV), de los Drs. Antonio García y Pedro Orizaola (Servicio de Neurofisiología Clínica, HUMV), del Dr. Javier Figols y de la Dra. Nuria Terán-Villagrá (Servicio de Anatomía Patológica, HUMV), de la Dra. Elena Gallardo (Servicio de Radiodiagnóstico, HUMV), y de los Prof. Miguel Lafarga y M.ª Teresa Berciano (Departamento de Anatomía y Biología Celular, Universidad de Cantabria). Agradezco al Dr. José Gazulla (Hospital Miguel Servet, Zaragoza) la revisión del manuscrito, y a Don Mario Corral (responsable de la Biblioteca Marquesa de Pelayo, Santander) su colaboración en la búsqueda de la bibliografía.

