



La enfermedad de Alzheimer (EA) es la principal enfermedad neurodegenerativa cortical. Su incidencia aumenta con la edad, lo que provoca importantes problemas médicos, sociales y económicos, especialmente en países con población envejecida.
ObjetivoEl objetivo de esta revisión es poner de manifiesto las evidencias que existen sobre el modo en que la disfunción vascular puede contribuir al deterioro cognitivo en la EA, así como las posibilidades terapéuticas que de ello podrían derivarse.
DesarrolloLa hipótesis vascular ha surgido como alternativa a la hipótesis de la cascada amiloide como explicación de la fisiopatología de la EA. Esta hipótesis sitúa en los vasos sanguíneos el origen de una serie de estímulos patogénicos que llevarían a la lesión neuronal y la demencia. La destrucción de la organización de la barrera hematoencefálica, la disminución del flujo sanguíneo cerebral y el establecimiento de un contexto inflamatorio serían responsables de un consecuente daño neuronal a causa de favorecer la agregación del péptido β-amiloide en el cerebro. Las vías que relacionan la disfunción vascular con la neurodegeneración han proporcionado nuevos enfoques terapéuticos y dianas farmacológicas con las que avanzar en la búsqueda de tratamientos para la EA.
ConclusionesResulta difícil determinar si el componente vascular de la EA es la causa o el efecto de la enfermedad, pero no cabe duda de que la enfermedad vascular tiene una relación importante con la EA. Es probable que la disfunción vascular actúe sinérgicamente con los cambios neurodegenerativos en un ciclo que agrava el deterioro cognitivo propio de la EA.
Alzheimer disease (AD) is the main cortical neurodegenerative disease. The incidence of this disease increases with age, causing significant medical, social and economic problems, especially in countries with ageing populations.
ObjectiveThis review aims to highlight existing evidence of how vascular dysfunction may contribute to cognitive impairment in AD, as well as the therapeutic possibilities that might arise from this evidence.
DevelopmentThe vascular hypothesis emerged as an alternative to the amyloid cascade hypothesis as an explanation for the pathophysiology of AD. This hypothesis locates blood vessels as the origin for a variety of pathogenic pathways that lead to neuronal damage and dementia. Destruction of the organisation of the blood brain barrier, decreased cerebral blood flow, and the establishment of an inflammatory context would thus be responsible for any subsequent neuronal damage since these factors promote aggregation of β-amyloid peptide in the brain. The link between neurodegeneration and vascular dysfunction pathways has provided new drug targets and therapeutic approaches that will add to the treatments for AD.
ConclusionsIt is difficult to determine whether the vascular component in AD is the cause or the effect of the disease, but there is no doubt that vascular pathology has an important relationship with AD. Vascular dysfunction is likely to act synergistically with neurodegenerative changes in a cycle that exacerbates the cognitive impairment found in AD.
La principal enfermedad neurodegenerativa cortical es la enfermedad de Alzheimer (EA), y su manifestación clínica más importante es la demencia, es decir, la pérdida progresiva de la función cognitiva1. El deterioro cognitivo en la EA suele seguir una evolución clínica característica que comienza con déficits de memoria que suelen pasar inadvertidos pero que poco a poco comienzan a interferir en las actividades cotidianas. En las etapas intermedias de la EA el paciente ya no puede trabajar, se pierde fácilmente, está confundido y necesita supervisión diaria. Aparecen déficits del lenguaje, en primer lugar en la asignación de nombres, después en la compresión y por último en la fluidez verbal. También surge apraxia y la persona muestra gran dificultad para realizar diferentes tareas motoras. En las etapas tardías de la enfermedad las personas vagan sin rumbo fijo, el razonamiento ha desaparecido y son frecuentes los delirios. En la fase terminal de la EA los pacientes están rígidos, mudos y necesitan ayuda para ejecutar sus funciones fisiológicas2.
Según el estudio mundial de Ferri et al.3, en la actualidad más de 23,4millones de personas sufren esta demencia y cada año aparecen 4,6millones de nuevos casos en el mundo, lo que equivale a un nuevo caso cada 7segundos. Por ello, se prevé que esta cifra se duplicará cada 20años, llegando a 81,1millones de personas afectadas en 2040. Los pacientes raramente desarrollan síntomas antes de la quinta década, pero la incidencia de la enfermedad aumenta con la edad. Este incremento progresivo ha dado lugar a problemas médicos, sociales y económicos, fundamentalmente en países con un número creciente de población envejecida1. En lo que se refiere a España, la prevalencia de la EA se ha estimado en un 7,7% en la población mayor de 70años, siendo su incidencia algo mayor en mujeres que en hombres4. En el año 2013, la EA se situó como la séptima causa de muerte y la primera entre las demencias en la población española5.
Dada la elevada prevalencia de la EA y las importantes implicaciones médicas, sociológicas y económicas que conlleva, el objetivo de este trabajo es poner de manifiesto las evidencias que existen actualmente sobre el modo en que la disfunción vascular puede contribuir al deterioro cognitivo en la EA, así como las posibilidades terapéuticas que de ello podrían derivarse.
La hipótesis de la cascada amiloide en la enfermedad de AlzheimerEn 1907, el psiquiatra Alois Alzheimer describió por primera vez los signos patognomónicos de la EA en August D., una mujer de 51años que había sido ingresada en un sanatorio mental con signos de demencia. En el examen histopatológico del cerebro de August D., Alzheimer identificó placas y ovillos neurofibrilares y afirmó que la demencia que padecía esta paciente estaba relacionada con las lesiones que observaba6,7.
Varios años después, en 1984, surgió la hipótesis de la cascada amiloide a raíz de que Glenner y Wong8 describieran que el componente fundamental de las placas observadas por Alzheimer era el péptido β-amiloide (Aβ). Tres años después, en 1987, Goldgaber et al.9 localizaron en el cromosoma 21 el gen que codificaba la proteína precursora del amiloide (APP) de la que procedía el péptido Aβ. Posteriormente, se descubrió que la EA podía ser heredada de forma autosómica dominante a partir de una mutación en esa misma proteína10. Este conjunto de observaciones articuló la hipótesis de la cascada amiloide con el ánimo de sintetizar la información histopatológica y genética que estaba entonces disponible para la EA. Dicha hipótesis fue desarrollada íntegramente en 1992 por Hardy y Higgings11 y actualizada en numerosas ocasiones desde entonces12.
Durante los años noventa se fueron acumulando un conjunto de evidencias experimentales que apoyaban decisivamente el papel de la APP y su procesamiento proteolítico como elementos claves en la fisiopatología de la EA. Se describieron distintas mutaciones relacionadas con la enfermedad en regiones de escisión proteolítica de la APP por parte de una serie de proteasas que se identificaron como α-, β- y γ-secretasas13. Asimismo, fue un hecho decisivo la demostración de cómo la generación del Aβ se producía al favorecer el procesamiento proteolítico de la APP por parte de la β-secretasa seguida de la acción de la γ-secretasa14,15. Por otra parte, en 1995 se había observado que las mutaciones autosómicas dominantes en el gen de la presenilina-1 (PTEN-1) y presenilina-2 (PTEN-2) presentes en el cromosoma 14 y 1, respectivamente, podían causar la EA16,17. Sin embargo, fueron Wolfe et al.18 en 1999 quienes demostraron que estas dos proteínas homólogas estructuran el sitio catalítico de la γ-secretasa y resultan esenciales para su función proteolítica. Esto último confirmaba decisivamente el papel de las secretasas y su actividad sobre la APP como elementos fundamentales en la fisiopatología de la EA.
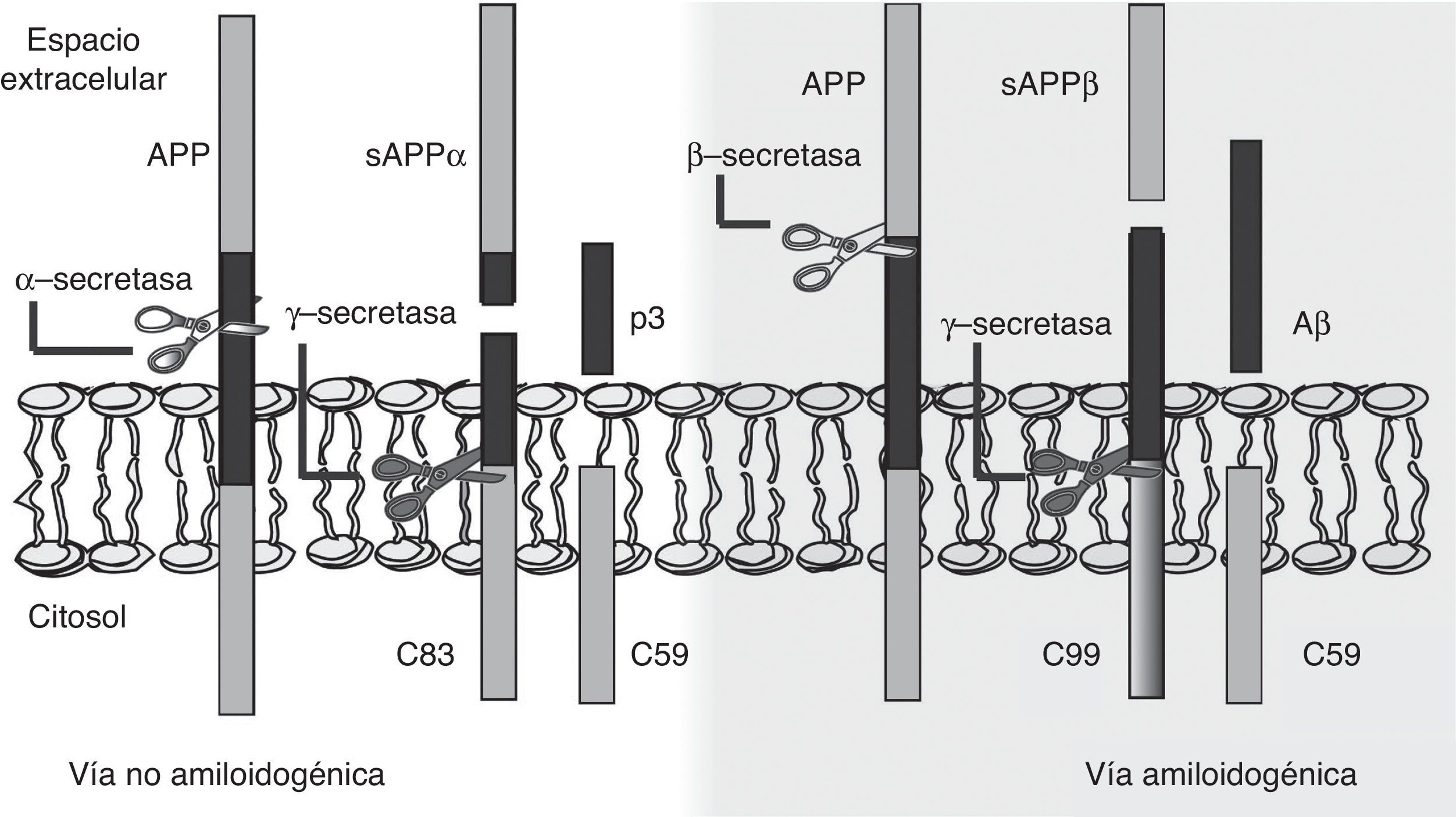
La hipótesis de la cascada amiloide señala, por tanto, que la producción del Aβ sería la responsable de la disfunción y muerte de las neuronas, lo que conduciría al estado neurodegenerativo y a la demencia19. Resulta clave, entonces, conocer el mecanismo que lleva a la generación del Aβ a partir de la APP, una proteína transmembrana de paso único de 770aminoácidos, con un gran dominio extracelular, expresada ampliamente en las neuronas pero cuya función fisiológica sigue sin estar totalmente esclarecida20. En este sentido, la hipótesis de la cascada amiloide plantea que el procesamiento de la APP puede ocurrir por 2 vías: bien por una vía no amiloidogénica o bien por una vía amiloidogénica (fig. 1). La primera de ellas es la más frecuente y se produce por la acción de la α-secretasa, que escinde la APP en el aminoácido83 desde el extremo C-terminal, generando un fragmento N-terminal que se secreta al medio extracelular (sAPPα), y un fragmento C-terminal de 83aminoácidos (C83) que se retiene en la membrana y que posteriormente es escindido por una γ-secretasa, produciendo un pequeño péptido soluble (p3). Sin embargo, si el procesamiento tiene lugar por la vía amiloidogénica, el primer corte proteolítico está mediado por la β-secretasa en una posición situada a 99aminoácidos del extremo C-terminal, resultando entonces la liberación de sAPPβ y reteniendo en la membrana un fragmento de 99aminoácidos (C99). A continuación, C99 es procesado por la γ-secretasa, que puede llevar a cabo el corte proteolítico en diferentes posiciones, liberando, como consecuencia, el péptido Aβ que puede contener entre 37 y 49aminoácidos (Aβ37–Aβ49), siendo Aβ40 el fragmento más frecuente. En cualquier caso, estos péptidos Aβ son capaces de agregarse en pequeñas estructuras oligoméricas, para finalmente formar las placas seniles típicas observadas en la EA21,22.
: vía no amiloidogénica y vía amiloidogénica. Esta última permite la generación del péptido β-amiloide (Aβ) y la formación de placas seniles.")
Actualmente, es bien conocido que el principal factor de riesgo genético para la EA es el gen de la apolipoproteína E (APOE), concretamente el alelo APOE423. Los seres humanos poseen 3 alelos comunes de APOE: APOE2, APOE3 y APOE4. El mecanismo por el cual las proteínas APOE median sus efectos en la EA no se conoce con exactitud, pero es posible que intervengan en la degradación del péptido Aβ de forma que APOE2, APOE3 y APOE4 sean, en este orden, menos eficaces para eliminar el Aβ24. Tiraboschi et al.25 demostraron la presencia de una mayor deposición de placas seniles en los cerebros de pacientes portadores de 2 alelos APOE4, mientras que la posesión del alelo APOE2 disminuía la presencia de dichas placas.
Alois Alzheimer observó, además de las placas seniles, la presencia de ovillos neurofibrilares como signo histopatológico de la EA6. La proteína tau hiperfosforilada es el componente principal de estas estructuras26, lo que ha generado que exista un creciente interés en conocer el papel de esta proteína en la EA. Se trata de una proteína asociada a los microtúbulos presente en la red microtubular de los axones neuronales. Con el desarrollo de la EA, tau se vuelve hiperfosforilada, lo que impide su capacidad de unirse a los microtúbulos y la estabilización de los mismos. Esta situación lleva a una reducción del transporte axonal y, con ello, a la disfunción y muerte neuronal. Probablemente exista una relación entre el péptido Aβ y la proteína tau, pero actualmente esta todavía no es conocida27.
La hipótesis vascular en la enfermedad de AlzheimerLa hipótesis vascular para explicar la patogénesis de la EA surge como una alternativa a la hipótesis de la cascada amiloide. Fue planteada por primera vez en 1993 por De la Torre y Mussivand28 al observar, en pacientes con EA, una reducción —proporcional a la gravedad de la enfermedad— en el flujo sanguíneo, en el metabolismo de la glucosa y en el consumo de oxígeno cerebral.
En la actualidad, la hipótesis vascular se apoya en una evidencia creciente que sugiere que la disfunción vascular juega un papel central en el desarrollo de la EA. Las placas seniles y los ovillos neurofibrilares podrían no ser la causa de la neurodegeneración, sino su consecuencia. En pacientes con traumatismo craneoencefálico se han observado estas estructuras histopatológicas, lo que sugiere que la sobreexpresión de la APP y la deposición amiloide podrían ocurrir en la fase de respuesta aguda al daño neuronal29. Además, el grado de deposición del péptido Aβ no se ha podido correlacionar con la gravedad de la disfunción cognitiva en los pacientes con EA, e incluso se han hallado deposiciones amiloides en cerebros de muchos individuos con funciones cognitivas normales30.
Son muchos los estudios que avalan la asociación entre los distintos factores de riesgo vascular y una mayor susceptibilidad de padecer EA. El estudio de Launer et al.31 concluyó que la hipertensión arterial aumentaba el riesgo de padecer EA en hombres de edades avanzadas que nunca habían sido tratados con antihipertensivos. Asimismo, Kivipelto et al.32 demostraron que el aumento de la presión arterial sistólica y los niveles elevados de colesterol sérico en personas de mediana edad aumentaban el riesgo de padecer EA en edades más avanzadas. La obesidad también ha sido vinculada a una mayor probabilidad de padecer EA. Muestra de ello es el estudio de Xu et al.33, cuyas conclusiones apoyan que tanto el sobrepeso como la obesidad, en edades medias, aumentan de forma independiente la predisposición a sufrir la EA. De entre los factores de riesgo de la EA asociados a la obesidad destaca la resistencia a la insulina34,35. De hecho, esta juega un papel clave en la fisiopatología de la diabetes mellitus tipo2, cuya relación con el riesgo de desarrollar EA ha sido avalada por distintos estudios epidemiológicos36,37. Por otra parte, la relación entre la aterosclerosis y la EA también ha quedado reflejada en estudios como en el de Hofman et al.38, que encontró una probabilidad 3 veces mayor de padecer EA en pacientes con aterosclerosis. Recientemente se ha contemplado también la posibilidad de que los factores de riesgo vascular puedan superponerse a los factores de riesgo genético vinculados con la EA. Rodrigue et al.39 abordaron la hipótesis de que los individuos con factores de riesgo vascular como la hipertensión, en combinación con un factor de riesgo genético para la EA (alelo E4 de la apolipoproteínaE), mostrarían una mayor carga de amiloide en el cerebro que aquellos individuos sin ese riesgo. En este estudio se encontró que el grupo de hipertensos con al menos un alelo E4 mostraron significativamente mayor deposición del Aβ que aquellos que solo tenían uno de los 2 factores de riesgo o no tenían ninguno.
Lesión en la unidad neurovascular en la enfermedad de AlzheimerLas células endoteliales, los pericitos, las neuronas y las células gliales forman, en conjunto, una unidad funcional conocida como «unidad neurovascular». La proximidad de los diferentes tipos de células no neuronales entre sí y con las neuronas permite regulaciones paracrinas que son críticas para la fisiología normal del sistema nervioso central (SNC). Así, las células vasculares, es decir, el endotelio y los pericitos, pueden afectar directamente a las funciones neuronales y sinápticas a través de las alteraciones en el flujo sanguíneo, en la permeabilidad de la barrera hematoencefálica (BHE), en el suministro de nutrientes, en la degradación eficiente de moléculas tóxicas, en las funciones enzimáticas, en la secreción de factores tróficos y moléculas de la matriz, en la expresión de receptores vasculares o en la inducción de la secreción de ectoenzimas40.
Dada la importante función que desarrolla la BHE en la regulación del paso de metabolitos circulantes al tejido cerebral, la disrupción de esta puede contribuir de forma importante a la acumulación de moléculas neurotóxicas en el cerebro. Se ha observado que los niveles de muchas proteínas de unión estrecha, adherente y/o de sus moléculas adaptadoras disminuyen en la EA, lo que conduce a una alteración de la permeabilidad de la BHE. Se ha demostrado, también, que en los trastornos neurodegenerativos se eleva la expresión de las metaloproteinasas de la matriz (MMP) vascular, de las que son sustrato las proteínas de unión estrecha y algunas proteínas de la matriz extracelular, cuya degradación podría explicar los cambios en la permeabilidad de la BHE en la EA41. La disfunción de la permeabilidad de la BHE ha sido explicada, además, por alteraciones en los sistemas de transporte selectivo que, mediados por moléculas transportadoras, facilitan el transporte de determinados nutrientes circulantes por la sangre hacia el interior del parénquima cerebral. Muestra de ello son estudios realizados en muestras de tejido cerebral de pacientes con EA donde existía una disminución en la expresión de GLUT-1, lo que reduciría el transporte de glucosa desde la sangre al parénquima cerebral y explicaría así el motivo de una disminución de los sustratos metabólicos necesarios para el normal funcionamiento neuronal42.
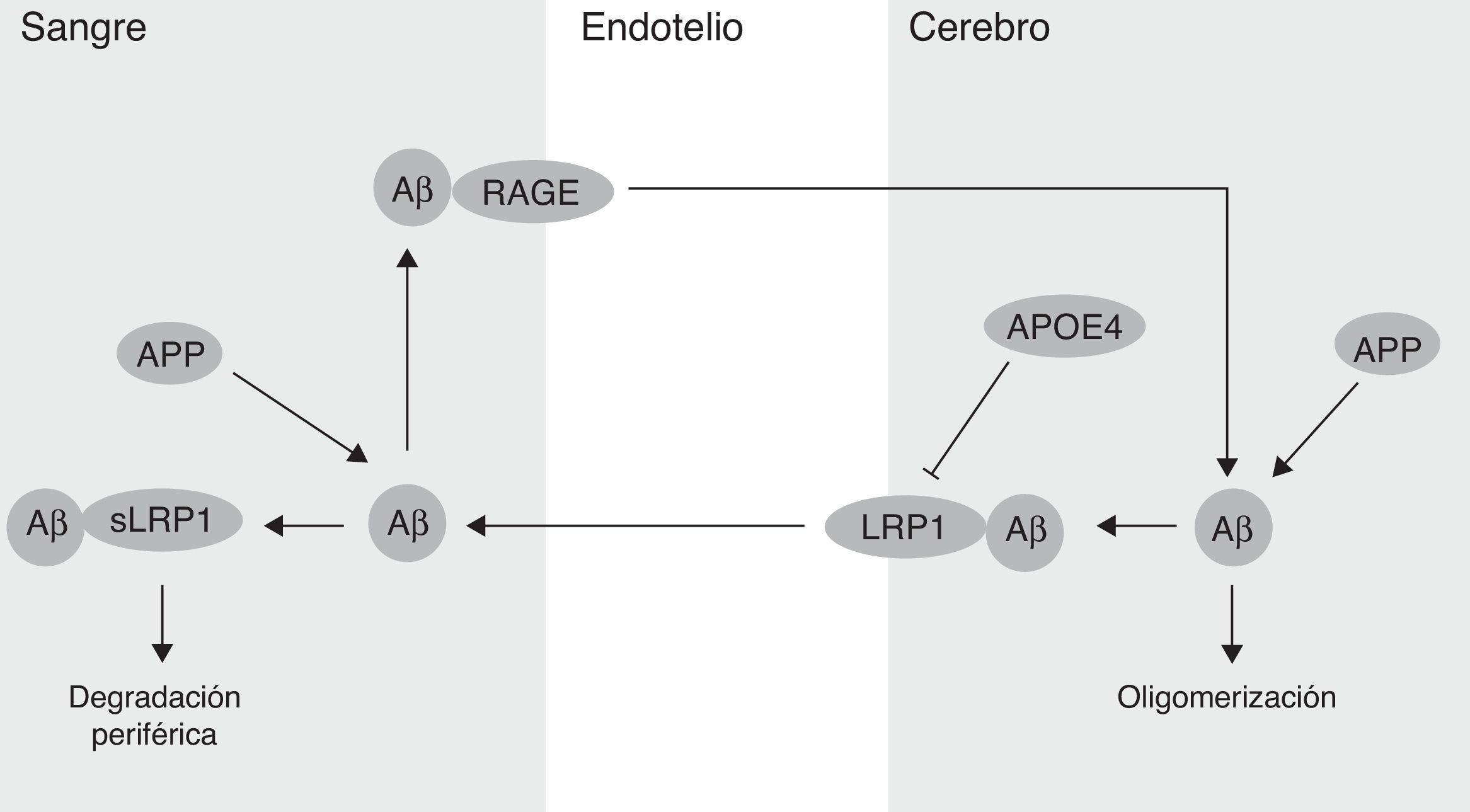
Por otra parte, se ha demostrado que la BHE participa en la regulación de los niveles del péptido Aβ en el SNC (fig. 2). De hecho, el Aβ periférico es un importante precursor del Aβ cerebral. Así, el receptor de los productos finales de glicación avanzada (RAGE) media el transporte del Aβ hacia el cerebro y la propagación de su toxicidad. La expresión del RAGE en el endotelio cerebral proporciona un mecanismo para la afluencia del péptido Aβ y de monocitos cargados con el Aβ a través de la BHE41. En modelos animales de la EA se ha demostrado que la expresión del RAGE está aumentada, amplificando de este modo las respuestas patogénicas mediadas por el péptido Aβ43. Por otra parte, varios estudios en modelos animales de la EA, y también en pacientes, han demostrado una disminución de la degradación del Aβ en el tejido cerebral afectado por esta enfermedad44,45. En este sentido, el receptor endotelial LRP1 juega un papel importante en la degradación del Aβ46. Los bajos niveles de LRP1 en los microvasos cerebrales están asociados con la acumulación del Aβ en el cerebro durante el envejecimiento y durante la EA47,48. Se ha demostrado también que el LRP1 cerebral se oxida en la EA mediante un mecanismo oxidativo que puede implicar al propio Aβ, lo que contribuye a la retención de este, debido a que la forma oxidada del LRP1 no puede unir y/o transportar el Aβ y dirigirlo hacia su degradación49. Finalmente, se ha estudiado que el APOE4, pero no el APOE3 ni el APOE2, bloquea el transporte mediado por el LRP1 del péptido Aβ desde el cerebro y, por lo tanto, promueve su retención50 (fig. 2). Además, se ha proporcionado evidencia de la relación existente entre el APOE, el LRP1 y el metabolismo del colesterol. La supresión del LRP1 en las neuronas del cerebro anterior de ratones adultos conseguía alterar significativamente los niveles cerebrales de APOE y de colesterol51.
Flujo sanguíneo cerebral y enfermedad de Alzheimer en el parénquima cerebral. APOE4: apolipoproteína E4; LRP1: receptor de la lipoproteína 1 de baja densidad; RAGE: receptor de los productos finales de glicación avanzada.")
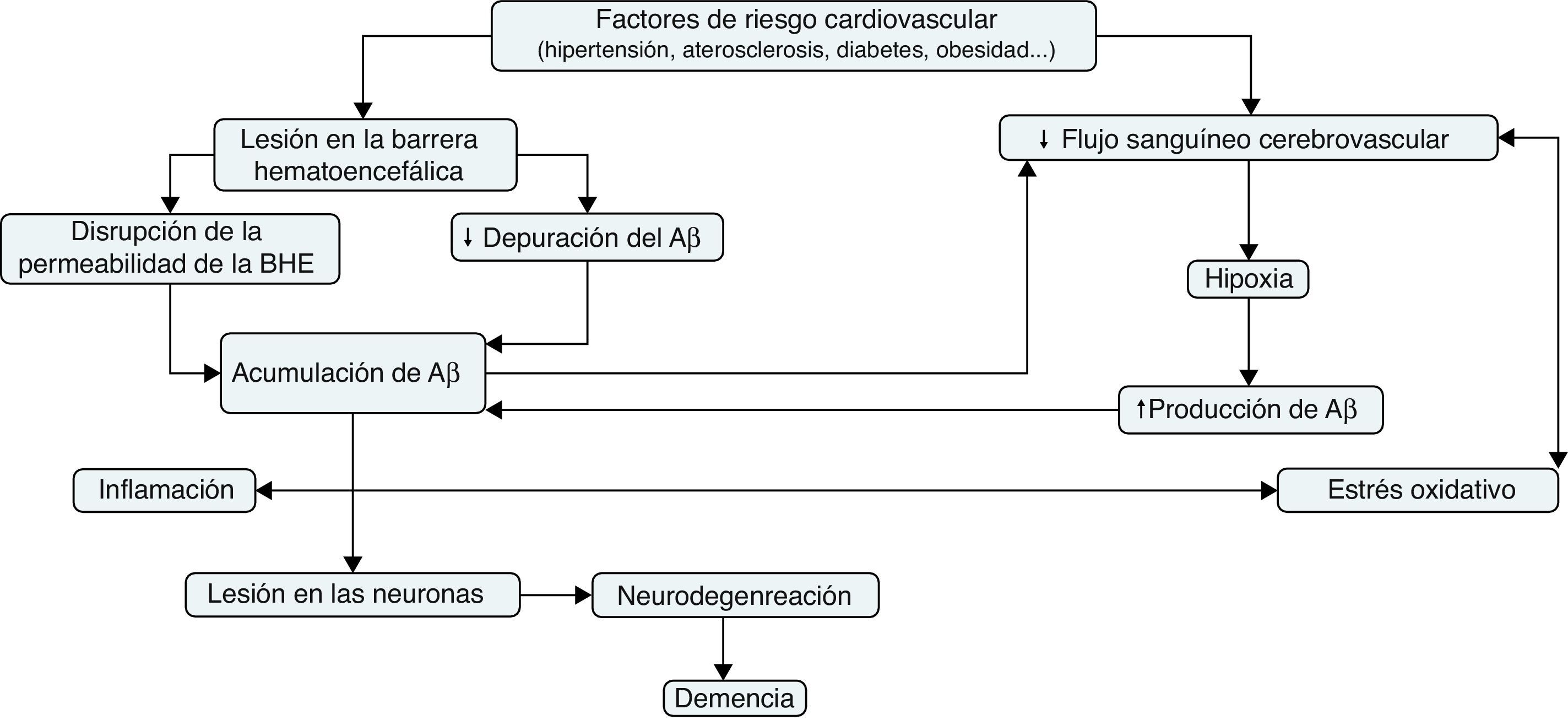
Una serie de resultados experimentales sugieren que la hipoperfusión cerebral precede al estado hipometabólico y neurodegenerativo propio de la EA52,53. La hipoperfusión cerebral crónica puede ser el resultado de la coexistencia de distintos factores de riesgo vascular, tales como la hipertensión, la diabetes, la aterosclerosis o las enfermedades cardiacas (fig. 3). Estos pueden afectar al sistema vascular cerebral y terminar produciendo una disminución progresiva del suministro de sangre al cerebro53,54. La disminución de la densidad microvascular, los vasos sanguíneos fragmentados y atróficos, el aumento de la irregularidad de los capilares, las modificaciones del diámetro de los vasos sanguíneos, el engrosamiento de la membrana basal de los capilares y la acumulación de colágeno en la misma son algunas disfunciones del tejido vascular que se han hallado en pacientes con EA55. Todo ello puede provocar lesiones neurodegenerativas, tales como la deposición de placas seniles u ovillos neurofibrilares, como resultado de un inadecuado suministro de sangre al cerebro53,54.

La hipoxia, resultante de la hipoperfusión cerebral, causa un aumento de la expresión del factor inducible por hipoxia (HIF)-1α en las neuronas. El HIF-1α se une al elemento sensible a la hipoxia del gen que codifica la β-secretasa, incrementando así la expresión del ARNm de la β-secretasa, y con ello la producción de fragmentos del Aβ56. Asimismo, se ha comprobado que la hipoxia crónica provoca una disminución de la expresión de la α-secretasa en las células del neuroblastoma humano57. Además, la hipoperfusión puede contribuir a una deficiencia de oxígeno que podría provocar estrés metabólico neuroglial, lo que haría aumentar más la producción del péptido Aβ y favorecería la producción de especies reactivas de oxígeno (ROS) mitocondriales58. Todas estas evidencias contribuyen a explicar los mecanismos moleculares que estarían detrás de los hallazgos que constatan que la hipoperfusión cerebral crónica acelera la deposición del Aβ59,60.
El propio péptido Aβ también puede influir en el flujo sanguíneo cerebral. Deane et al.61 demostraron que la administración sistémica del Aβ interacciona con el receptor RAGE en la pared de los vasos sanguíneos. Como resultado de dicha interacción, además de conducir el péptido Aβ al interior del parénquima cerebral, también favorece la producción de endotelina-1, un factor vasoconstrictor derivado del endotelio.
El estrés oxidativo vascular se ha convertido, también, en un factor patogénico clave en la EA, relacionado con la disfunción del endotelio y la disminución del flujo sanguíneo cerebral (fig. 3). El estrés oxidativo provoca disfunción endotelial a causa de la reacción del superóxido con el óxido nítrico (NO) endotelial para generar peroxinitrito, un oxidante potencialmente perjudicial que puede provocar daño oxidativo a las proteínas, los lípidos y los ácidos nucleicos. La pérdida de la biodisponibilidad de NO a causa de esta reacción provoca una disminución de la vasodilatación mediada por este. Por otra parte, la tetrahidrobiopterina —un cofactor crítico para las NO sintasas— es objeto de la oxidación mediada por el peroxinitrito y, en su defecto, estas enzimas se desacoplan, produciendo más ROS en lugar de NO, lo que causa más estrés oxidativo y dificulta la vasodilatación58. En general, el papel del NO en la fisiopatología de la EA es cada vez más relevante. Austin et al.62 demostraron que la inhibición, mediante NG-nitro-L-arginina metiléster (L-NAME), de la óxido nítrico sintasa endotelial (eNOS), enzima responsable de la síntesis del NO endotelial a partir de L-arginina, condujo a un aumento de los niveles de expresión de la proteína APP y de la β-secretasa en las células endoteliales de los microvasos del cerebro humano. Asimismo, el tejido cerebral de ratones eNOS mostraba niveles superiores de las proteínas APP y β-secretasa y un aumento de la actividad enzimática de esta última, en comparación con los ratones de fenotipo salvaje. Estos datos sugieren que el NO endotelial juega un papel importante en la modulación de la expresión y el procesamiento proteolítico de la APP, tanto en el cerebro como en los vasos sanguíneos cerebrales.
Como resumen de lo expuesto, podría afirmarse que la hipoperfusión cerebral favorece la acumulación del péptido Aβ en el parénquima cerebral y que, a su vez, la presencia del Aβ agrava la enfermedad vascular. Esta última podría actuar sinérgicamente con los cambios en los niveles del Aβ mediante un sistema de retroalimentación positiva, según el cual el daño tisular producido por los factores vasculares podría aumentar el daño producido por la neurodegeneración, y viceversa.
Inflamación, disfunción vascular y enfermedad de AlzheimerLas alteraciones en las funciones metabólicas cerebrovasculares pueden también conducir a la secreción de múltiples factores inflamatorios. En comparación con los controles, en los microvasos cerebrales de individuos con EA hay un aumento de los niveles del factor de necrosis tumoral (TNF), interleucina-1β (IL-1β) e IL-6, quimiocinas (CCL2 y IL-8), MMP y moléculas de adhesión de leucocitos63. Existe, además, evidencia que indica que distintos mediadores de la inflamación son capaces de regular la producción del Aβ (fig. 3). Zhao et al.64 observaron que la estimulación de astrocitos mediante citoquinas inflamatorias tales como TNF-α e interferón-γ (INF-γ) aumentaba los niveles de la APP, la β-secretasa y la secreción del Aβ. Por otra parte, se ha demostrado que la exposición de las células endoteliales del cerebro al péptido Aβ provoca una serie de respuestas proinflamatorias. El péptido Aβ, a través de su interacción con el receptor RAGE, regula la expresión de la quimiocina CCR5 y promueve la migración de las célulasT a través de la BHE65. Además, las células endoteliales cerebrales expuestas in vitro al péptido Aβ sobreexpresan genes inflamatorios, tales como MCP-1, IL-1β e IL-6 y aumentan la producción de prostaglandinas63.
En general, todo lo expuesto proporciona un enlace molecular entre la disfunción vascular, la lesión neuronal y la inflamación en la EA. En este sentido, es posible considerar que la microvasculatura cerebral participa de un ciclo destructivo en el que la inflamación podría preceder a la deposición del Aβ y, a su vez, el Aβ promover la liberación de mediadores inflamatorios.
Aproximaciones terapéuticas en la enfermedad de Alzheimer basadas en la hipótesis vascularTeniendo en cuenta la evidencia actual que sugiere que la hipoperfusión cerebral y la inflamación son importantes en el desarrollo de la EA, el uso de medicamentos tales como los antiinflamatorios no esteroideos (AINE), las estatinas y los antihipertensivos se han considerado prometedores para la prevención o el tratamiento de la EA58,66.
En el caso de los AINE, una serie de estudios epidemiológicos retrospectivos sugieren que estos fármacos son capaces de reducir significativamente el riesgo de desarrollar la EA67,68, corroborando así los ensayos en modelos animales de la EA en los que la terapia antiinflamatoria reducía la deposición cerebral del Aβ69. Sin embargo, los ensayos clínicos prospectivos para el estudio del efecto terapéutico de los AINE no han demostrado beneficio alguno70,71. La inconsistencia de estos resultados puede deberse al hecho de que los estudios epidemiológicos se realizan en pacientes en los que las manifestaciones clínicas de la enfermedad todavía no son evidentes, mientras que los ensayos clínicos se diseñan con personas cuyos síntomas ya son, en ese momento, clínicamente detectables. Además, dado el gran número de fármacos antiinflamatorios y la diversidad en sus actividades farmacológicas, es esencial optimizar la selección de los medicamentos en el diseño de los estudios clínicos. Para lograr este objetivo es fundamental una mejor comprensión de los mecanismos que median la influencia de los procesos inflamatorios en la progresión de la EA72.
En lo que respecta a las estatinas, los ensayos clínicos no han demostrado la eficacia de estos fármacos ni en la prevención ni en el tratamiento de la EA73–75. Los datos experimentales en modelos animales sí avalaron que las estatinas podrían prevenir la formación de placas seniles. Sin embargo, no existía evidencia que indicase que estos fármacos eran capaces de reparar las neuronas dañadas o de degradar las placas seniles ya existentes76,77. Es posible que los ensayos clínicos se hayan realizado en un momento en el que ya se había perdido la oportunidad terapéutica, interviniendo demasiado tarde, cuando el daño ya era irreversible o no quedaba nada que preservar78.
En lo referente a la medicación antihipertensiva, Forette et al.79 demostraron en un ensayo clínico que el tratamiento antihipertensivo se asociaba con una menor incidencia de la demencia. En el caso concreto de la EA, Yasar et al.80 mostraron que el uso de diuréticos, de bloqueadores de los receptores de la angiotensina-1 y de inhibidores de la enzima convertidora de la angiotensina se asociaba con un menor riesgo de desarrollar la EA en pacientes con cognición normal. No obstante, solo el uso de diuréticos se pudo asociar con un menor riesgo en los pacientes con deterioro cognitivo leve. Actualmente, continúa debatiéndose la idoneidad de utilizar antihipertensivos en el tratamiento de la EA, y se requiere una mayor cantidad de ensayos clínicos para confirmar su valor terapéutico real81.
Por otra parte, el creciente conocimiento de la fisiopatología de la EA y su relación con la disfunción vascular ha permitido identificar novedosas dianas terapéuticas. La observación de todas las respuestas mediadas por el receptor RAGE en el desarrollo de la EA ha propiciado que el bloqueo de este receptor sea una estrategia prometedora en el tratamiento de la EA. Asimismo, el LRP1 podría ser una nueva diana terapéutica para la EA. El LRP1 soluble circulante en el plasma se une al Aβ plasmático y dirige su degradación. Es por ello que fragmentos del LRP1 pueden tener un gran potencial terapéutico como agentes depuradores del péptido Aβ en la EA82. En la actualidad, todavía no hay datos clínicos suficientes como para validar el potencial terapéutico de estas nuevas estrategias83. Únicamente un estudio clínico reciente ha aportado cierta evidencia de que un inhibidor del RAGE a bajas dosis es capaz de frenar el deterioro cognitivo en pacientes con EA84. En cualquier caso, el estudio del modelo neurovascular de la EA ha proporcionado, y es esperable que continúe haciéndolo, nuevos mecanismos moleculares sobre los que poder intervenir farmacológicamente en el tratamiento de la EA.
ConclusionesEl modelo neurovascular de la EA propone que múltiples vías patogénicas provenientes de los vasos sanguíneos cerebrales podrían ser el estímulo patológico inicial. Este punto de vista es contrapuesto a la visión tradicional que considera las neuronas como el origen de la EA y que entiende que la enfermedad vascular subyacente es secundaria a la neurodegeneración. En cualquier caso, la evidencia disponible parece consensuar que la enfermedad vascular podría actuar sinérgicamente con los cambios neurodegenerativos, lo que resultaría en un deterioro cognitivo mayor.
En realidad resulta difícil determinar con exactitud si el componente vascular de la EA es la causa o el efecto de la enfermedad, pero no cabe duda que la enfermedad vascular tiene una relación importante en la progresión de la EA y se halla asociada a la disfunción neuronal. El nexo común entre los factores de riesgo vascular y la neurodegeneración pasaría por la disrupción de la BHE y la disminución del flujo sanguíneo cerebral. La destrucción de la BHE favorecería la acumulación del péptido Aβ y disminuiría la eficacia de la eliminación de este fragmento peptídico. Por su parte, la disminución del flujo sanguíneo cerebral aumenta la expresión de la APP y la actividad de la β-secretasa, lo que resulta en una acumulación mayor del Aβ. A partir de aquí, se establecería un mecanismo de retroalimentación positiva según el cual la hipoperfusión cerebral favorecería la acumulación del Aβ y, a su vez, la presencia del Aβ agravaría la disfunción vascular, causaría inflamación y estrés oxidativo. Todo ello aceleraría la neurodegeneración y provocaría los déficits cognitivos propios de la EA.
Considerar la influencia que el sistema vascular tiene sobre el desarrollo de esta enfermedad abre la puerta al descubrimiento de nuevas dianas terapéuticas y proporciona una nueva perspectiva a la hora de enfocar el mejor tratamiento de la EA. Por lo tanto, resulta fundamental el estudio de la contribución vascular a la fisiopatología de la EA.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.

