



Asymptomatic/paucisymptomatic hyperCKaemia is a frequent reason for consultation at neuromuscular disorders units. Elevated creatine kinase (CK) levels, or hyperCKaemia, may be caused by multiple factors, including hereditary myopathies. One such hereditary myopathy is anoctaminopathy secondary to mutations in the ANO5 gene (11p14.3). Recessive ANO5 mutations cause a wide range of clinical phenotypes, including limb-girdle muscular dystrophy type 2 L, Miyoshi myopathy 3, and isolated hyperCKaemia or hyperCKaemia associated with exercise intolerance.1
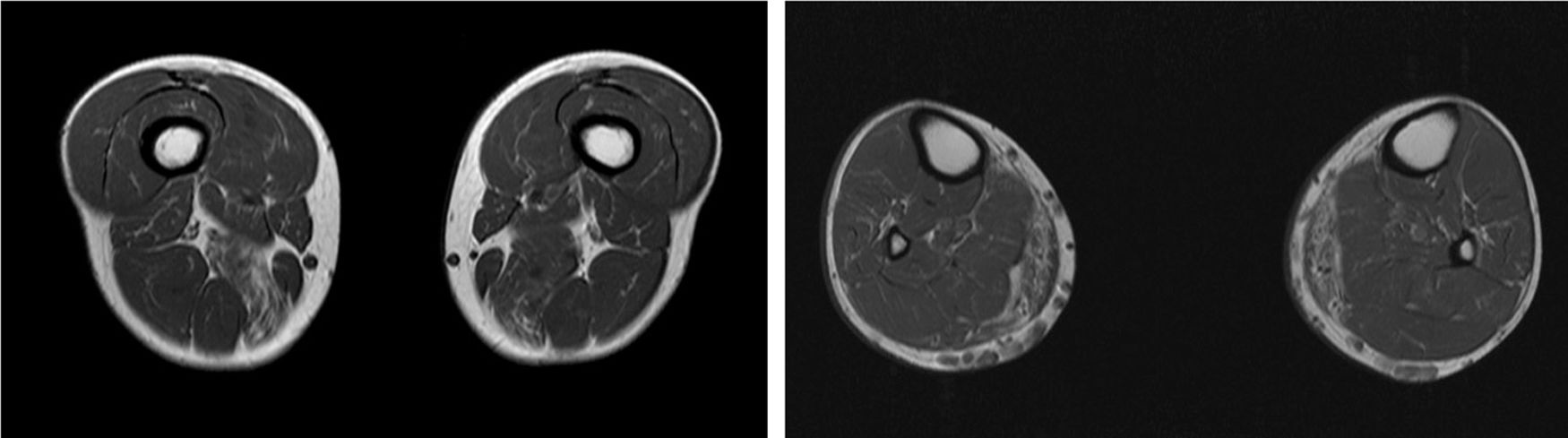
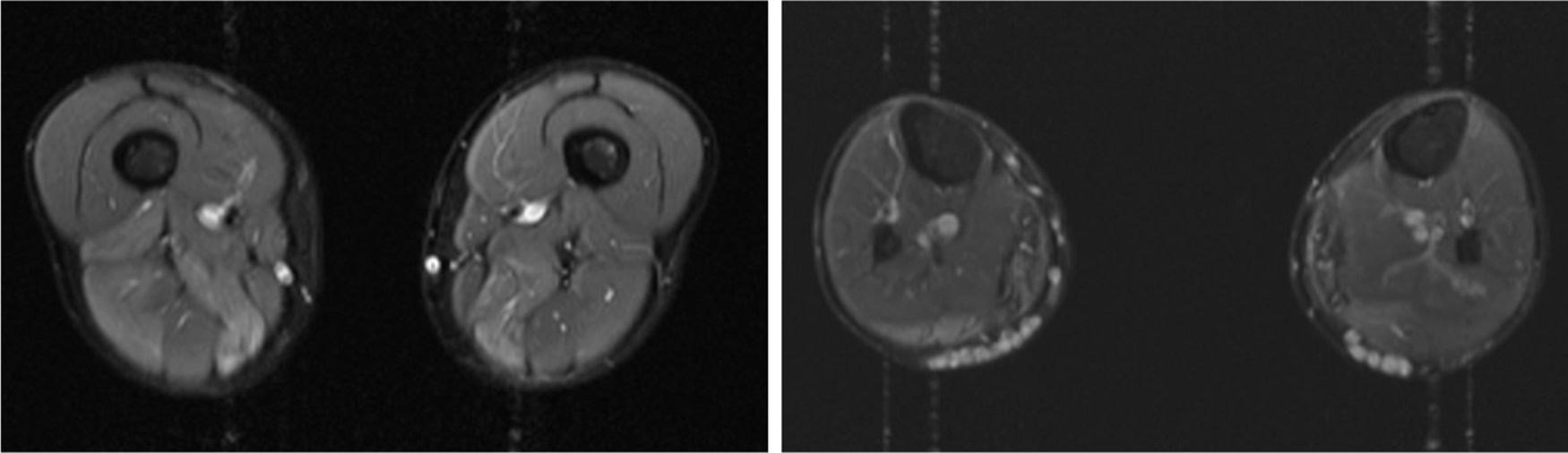
We present the case of a 41-year-old man with no relevant medical history except for hyperCKaemia of 6 years’ progression, detected in a routine blood test (1200–4500 U/L). He practised both aerobic and anaerobic exercise, and had developed exercise intolerance over the previous year. He had never presented choluria or rhabdomyolysis. The examination revealed no muscle weakness, amyotrophy, or myotonia. An electromyography study showed motor unit potentials with myopathic features in the muscles of both lower limbs, with no pathological spontaneous activity. A muscle MRI scan revealed asymmetric fatty infiltration in the semimembranosus and medial gastrocnemius muscles (Fig. 1), with myoedema in the semimembranosus, biceps femoris, and gastrocnemius muscles on STIR sequences (Fig. 2), despite the lack of clinical evidence of weakness. A transthoracic echocardiogram and Holter electrocardiography found no alterations. A muscle biopsy revealed a dystrophic pattern, with variations in fibre size, marked nuclear internalisation, and signs of necrosis and regeneration. An immunohistochemical study and Western blot analysis detected no alterations in dystrophin, sarcoglycans, calpain, dysferlin, or caveolin.


In view of the nonspecific dystrophic alterations revealed by the MRI and biopsy studies, we requested a next-generation sequencing analysis of a panel of genes associated with neuromuscular disorders. The analysis identified a homozygous variant, c.1982 T > C, in exon 8 of ANO5 (reference sequence: NM_213599), which causes amino acid substitution p.Leu661Pro. This variant is registered in the gnomAD database with a frequency of 0.00003230, but had not previously been associated with any disease. A genetic study of the patient’s relatives revealed that both parents were heterozygous carriers of the mutation. After over 8 years of follow-up, the patient has developed no further symptoms besides mild exercise intolerance.
We present a case of paucisymptomatic hyperCKaemia secondary to a mutation in the ANO5 gene; diagnosis was reached with massive DNA sequencing of a pre-established group of genes associated with our patient’s symptoms. As mentioned previously, the aetiology of hyperCKaemia is multifactorial. Several diagnostic algorithms can be used, which include different complementary tests2,3; gene panel sequencing is not currently included. Among the initial tests performed in our patient, muscle MRI and biopsy results suggested an underlying dystrophy, but not the specific type. Muscle MRI revealed a pattern compatible with anoctaminopathy, given the asymmetric involvement of the posterior compartment of the thigh and calf.4 However, this finding is nonspecific, and such other complementary tests as muscle biopsy are required. Although amyloid deposition5 and necrotising myopathy6 have been observed in patients with anoctaminopathy, no histopathological markers currently available for use in muscle biopsy provide diagnostic certainty. In view of the above, we requested next-generation sequencing of a panel of genes, reaching a definitive diagnosis. Our results are consistent with those from recent studies into patients with asymptomatic/paucisymptomatic hyperCKaemia, many of whom presented ANO5 mutations.7
DNA sequencing technologies have evolved considerably in recent years; the full genome can be sequenced rapidly and at relatively low cost thanks to such techniques as next-generation sequencing.8 Due to the considerable genetic heterogeneity of neuromuscular disorders, a comprehensive study of all possible mutations is not always possible. Given that molecular diagnosis must take clinical factors into consideration, analysing panels of genes associated with a specific condition, such as hyperCKaemia in our patient, seems to be the most cost-effective strategy.9 In the light of the above, it seems reasonable to include gene panel sequencing in the diagnostic algorithm for asymptomatic/paucisymptomatic hyperCKaemia, particularly when the results of previous tests (eg, MRI, muscle biopsy) suggest an underlying myopathy.
Please cite this article as: Alcahut-Rodríguez C, Díaz-Maroto I, Fernandez-Marmiesse A, García-García J. HiperCKemia paucisintomática secundaria a mutación en el gen ANO5. Neurología. 2020;35:510–512.
