



Pyramidal signs (hyperreflexia, spasticity, Babinski sign) are essential for the diagnosis of amyotrophic lateral sclerosis (ALS). However, these signs are not always present at onset and may vary over time, besides which their role in disease evolution is controversial. Our goal was to describe which pyramidal signs were present and how they evolved in a cohort of patients with ALS, as well as their role in prognosis.
MethodsRetrospective analysis of prospectively collected patients diagnosed with ALS in our centre from 1990 to 2015.
ResultsOf a total of 130 patients with ALS, 34 (26.1%) patients showed no pyramidal signs at the first visit while 15 (11.5%) had a complete pyramidal syndrome. Of those patients without initial pyramidal signs, mean time of appearance of the first signs was 4.5 months. Babinski sign was positive in 64 (49.2%) patients, hyperreflexia in 90 (69.2%) and 22 (16.9%) patients had spasticity. Pyramidal signs tended to remain unchanged over time, although they seem to appear at later stages or even disappear with time in some patients.
We found no association between survival and the presence of changes to pyramidal signs, although decreased spasticity was associated with greater clinical deterioration (ALSFR scale) (P<.001).
ConclusionA quarter of patients with ALS initially showed no pyramidal signs and in some cases they even disappear over time. These data support the need for tools that assess the pyramidal tract.
Los signos piramidales (hiperreflexia, espasticidad, signo de Babinski) son fundamentales para el diagnóstico de esclerosis lateral amiotrófica (ELA). Sin embargo, no siempre están presentes al comienzo, pueden variar con el tiempo y es controvertido su papel en la evolución. El objetivo del estudio es describir qué signos piramidales están presentes inicialmente y cómo evolucionan en una cohorte de pacientes con ELA, así como su papel pronóstico.
MétodosAnálisis retrospectivo de pacientes recogidos de manera prospectiva, diagnosticados de ELA en nuestro centro, desde 1990 hasta 2015.
ResultadosDel total de 130 pacientes con ELA, 34 (26,1%) no presentaron inicialmente ningún signo piramidal, mientras que 15 (11,5%) presentaban un síndrome piramidal completo. De aquellos pacientes sin piramidalismo inicial, la mediana de aparición de los primeros signos fue de 4,5 meses. El signo de Babinski estaba presente en 64 (49,2%), la hiperreflexia en 90 (69,2%) y en 22 (16,9%) pacientes existía espasticidad. Los signos piramidales tendían a mantenerse inalterados en el tiempo, aunque existe un porcentaje de pacientes en el que aparecen tardíamente o desaparecen con el tiempo.
No se encontró asociación entre supervivencia y la presencia o modificación de signos piramidales, aunque la disminución de la espasticidad se asociaba a mayor deterioro clínico (escala ALSFR) (p<0,001).
ConclusiónUna cuarta parte de pacientes con ELA no presentaron inicialmente ningún signo piramidal y, en algunos casos, estos desaparecen con el tiempo. Esto resalta la necesidad de la inclusión de herramientas para la valoración de la vía piramidal.
Combined degeneration of the upper and lower motor neurons constitutes the pathological hallmark of amyotrophic lateral sclerosis (ALS).1 The diagnosis of ALS requires evidence of lower motor neuron damage from clinical or neurophysiological studies,2 combined with evidence of upper motor neuron damage, which is determined by clinical examination. Pyramidal signs are not always present at disease onset, however, and may change over the course of the disease. Assessment of these signs may be subject to inter- and intraexaminer variations. Furthermore, their role in disease progression and prognosis is yet to be determined.3,4
The purpose of this study is to conduct a descriptive analysis of the presence of pyramidal signs in a cohort of patients with ALS, both at baseline and throughout the course of the disease, in addition to studying the association between pyramidal signs and prognosis.
MethodsWe conducted a retrospective analysis of patients diagnosed with motor neuron disease at our centre between 1991 and 2015, using a prospective database. The analysis included those patients meeting the revised El Escorial diagnostic criteria2 for probable or definite ALS and gathered data on clinical, epidemiological, and functional (ALSFRS scores at 6 and 12 months) variables and survival data (from symptom onset to death). We excluded those patients with pure upper (primary lateral sclerosis) or lower motor neuron disease (progressive muscle atrophy) and those for whom no data on disease progression were available. The following variables related to pyramidal signs were recorded: muscle tone (increased, decreased, normal), deep tendon reflexes (hyperactive, present, diminished/absent), and plantar reflex (extensor, flexor, indifferent); pyramidal signs were evaluated at baseline and at every follow-up consultation (mean of 3 months between visits) until the last consultation (before the patient died or the study ended). Pyramidal signs were considered to be present when they affected at least 2 limbs, in the case of hypertonia and hyperreflexia, or at least one limb, in the case of the Babinski sign. Changes in a pyramidal sign were recorded as such when they were observed in at least 2 consecutive visits. Neurological examinations were performed by 2 expert neurologists. The study was approved by the ethics committee at our centre, Hospital del Mar, in Barcelona.
Statistical analysisComparisons between 3 groups were conducted using the chi-square test for categorical variables and the Kruskal-Wallis test for quantitative variables. Comparisons between 2 groups were conducted using the Mann-Whitney U test for quantitative variables and the Fisher exact test for categorical variables. Survival times were analysed using the Kaplan-Meier method; Kaplan-Meier curves were compared with the log-rank test. The Cox regression model was used to conduct a multivariate survival analysis which included the variables previously shown to cause statistically significant differences in survival time in the Kaplan-Meier analysis (P<.05). Data were analysed using SPSS statistical software, version 19.0. The end date for data inclusion was 1 May 2015.
ResultsOf a total of 256 patients with motor neuron disease, we excluded 40 with progressive muscular atrophy, 12 with primary lateral sclerosis, and 74 for whom no data on clinical progression were available. Our final sample included 130 patients diagnosed with ALS. Of these, 67 (51.5%) were men and 63 (48.5%) were women; mean age in our sample was 66 years. Disease onset was bulbar in 49 patients (37.7%) and spinal in 81 (62.3%). A total of 112 patients (86.1%) were deceased at the time of the study, with a median survival time of 29 months.
Functional status was measured with the ALSFRS. The median score was 37 points at baseline; scores decreased by 5 and 14 points at 6 and 12 months, respectively, from disease onset.
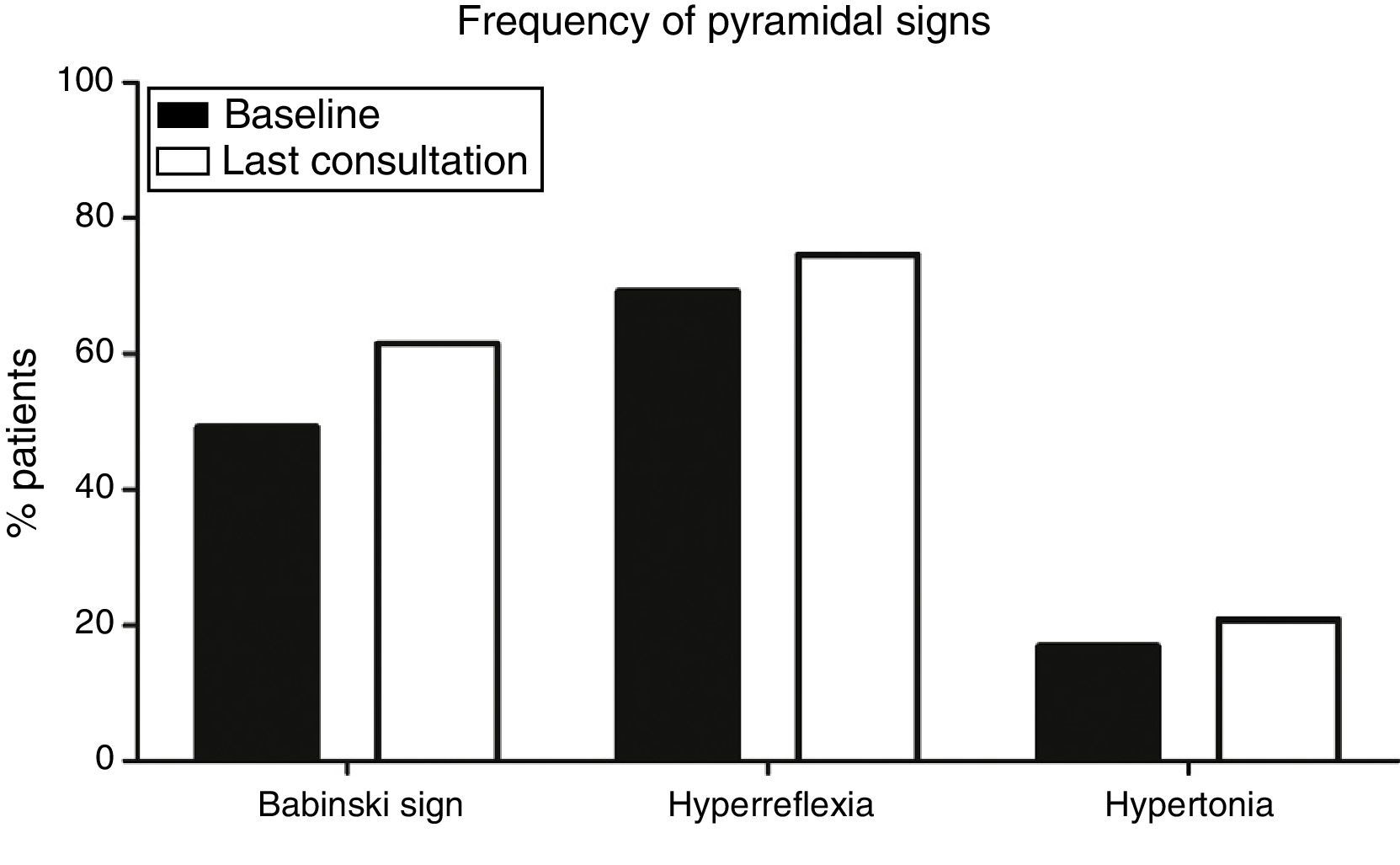
Pyramidal signs at baselineOf a total of 130 patients, baseline examination found that 15 (11.5%) had pyramidal syndrome, that is, Babinski sign, hyperreflexia, and increased muscle tone, whereas 34 (26.1%) displayed no pyramidal signs (median time of onset of the first pyramidal sign was 4.5 months). At baseline, Babinski sign (64 patients, 49.2%), and hyperreflexia (90, 69.2%), were more frequent than spasticity (22 patients, 16.9%) (P<.001) (Fig. 1).
 and hyperreflexia (69.2% at onset, 74.6% at the last consultation) were significantly more frequent (P<.001) than hypertonia (16.9% at onset, 20.8% at the last consultation).")
Frequency of pyramidal signs at baseline and at the last consultation. Babinski sign (49.2% at onset; 61.5% at the last consultation) and hyperreflexia (69.2% at onset, 74.6% at the last consultation) were significantly more frequent (P<.001) than hypertonia (16.9% at onset, 20.8% at the last consultation).
No statistically significant differences were found in sex, form of onset (spinal/bulbar), or survival time between patients with and without any of the pyramidal signs, or between those with or without pyramidal syndrome at baseline.
Progression of pyramidal signsPatients with ALS will invariably display pyramidal signs at some point during disease progression. This is a necessary condition for diagnosis. Over the course of the disease, however, some signs may disappear: in our sample, 12 patients (9.2%) showed no pyramidal signs at the last consultation and only 24 patients (18.5%) displayed all 3 pyramidal signs at the last consultation. Again, Babinski sign (80 patients, 61.5%) and hyperreflexia (97, 74.6%) were more frequent at the last consultation than hypertonia (27, 20.8%) (P<.001) (Fig. 1).
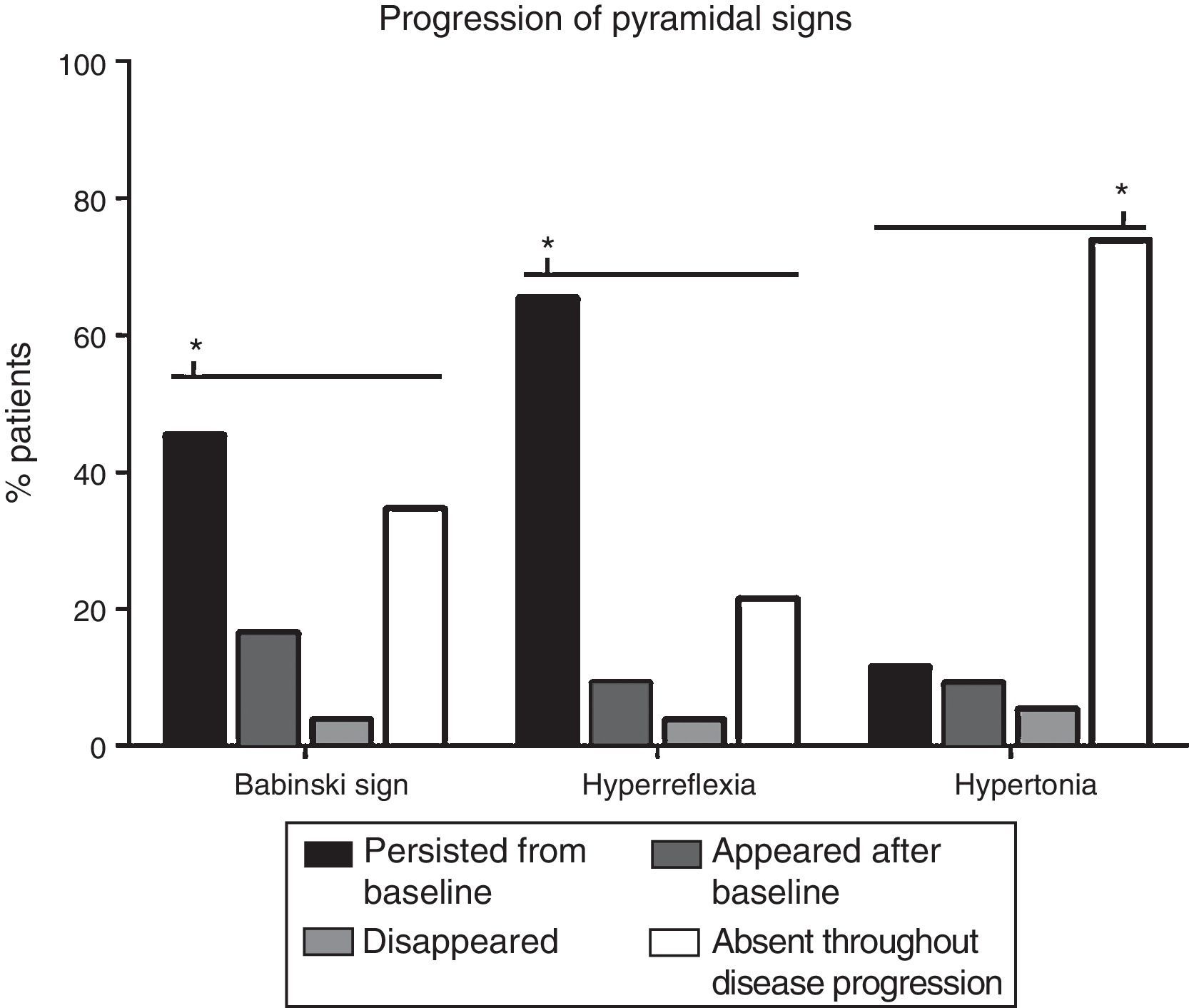
The progression of pyramidal signs is shown in Fig. 2. The most frequent progression observed both for hyperreflexia (65.4%) and for Babinski sign (45.4%) was persistence over the course of the disease (P<.0001). The most frequent finding for muscle tone was a lack of spasticity throughout the course of the disease (73.8%) (P<.0001).
 (P<.0001). Regarding muscle tone, spasticity remained absent throughout disease progression in most patients (73.8%) (P<.0001). Babinski sign appeared after the baseline consultation in 21 patients (16.5%), disappeared in 5 (3.8%), and remained absent in 45 (34.6%). Hyperreflexia appeared after baseline in 12 patients (9.2%), disappeared in 5 (3.8%), and remained absent in 28 (21.5%). Spasticity appeared after baseline in 12 patients (9.2%), disappeared in 7 (5.4%), and persisted in 15 (11.5%). *P<.0001.")
Analysis of changes in pyramidal signs throughout the course of the disease. The most frequent progression for hyperreflexia and Babinski sign was persistence from baseline (65.4% and 45.4%, respectively) (P<.0001). Regarding muscle tone, spasticity remained absent throughout disease progression in most patients (73.8%) (P<.0001). Babinski sign appeared after the baseline consultation in 21 patients (16.5%), disappeared in 5 (3.8%), and remained absent in 45 (34.6%). Hyperreflexia appeared after baseline in 12 patients (9.2%), disappeared in 5 (3.8%), and remained absent in 28 (21.5%). Spasticity appeared after baseline in 12 patients (9.2%), disappeared in 7 (5.4%), and persisted in 15 (11.5%). *P<.0001.
No differences were found in sex, age at onset, form of onset, or survival time between patients in whom pyramidal signs changed and those in whom signs remained stable.
Pyramidal signs and prognosisNo statistically significant differences were found in survival times between patients with and without any pyramidal sign at baseline or between patients with and without changes in pyramidal signs over the course of the disease. Likewise, we observed no association between survival time and presence of pyramidal syndrome or absence of all pyramidal signs at baseline.
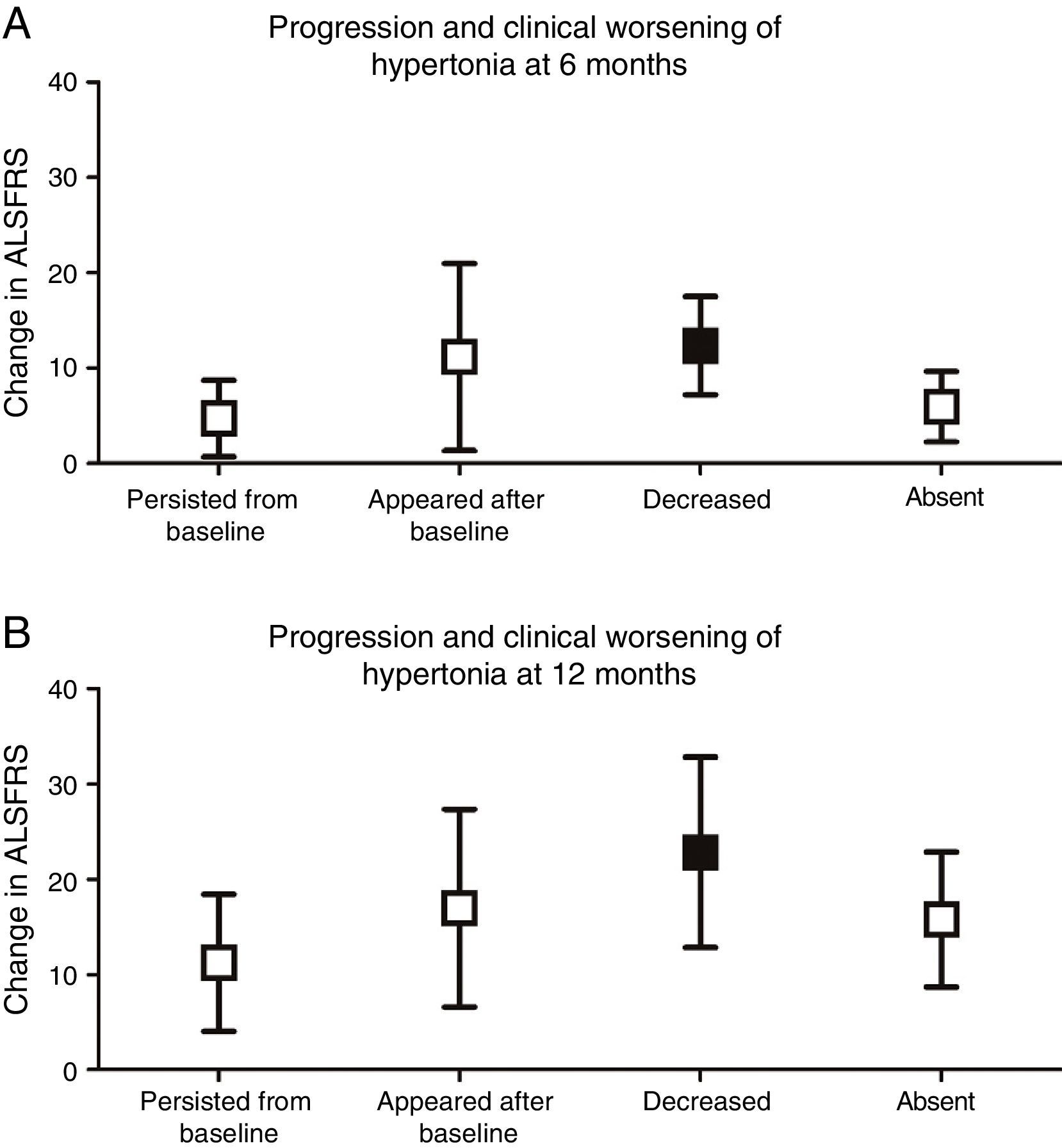
We analysed whether any of the different types of progression of each pyramidal sign were associated with greater functional impairment. Only complete resolution of spasticity was found to be associated with more severe clinical deterioration at 6 (P<.001) and 12 months (P<.01), compared to baseline ALSFRS scores (Fig. 3).
. (A) At 6 months, clinical worsening was more marked in patients in whom hypertonia decreased over the course of the disease (12.33±5.16) (P<.001) than in those in whom spasticity persisted (4.69±4.0), appeared after baseline (11.14±9.81), or remained absent (5.92±3.69). (B) At 12 months, clinical worsening was also more marked in those patients in whom hypertonia disappeared (22.83±9.99) (P<.01) than in those in whom hypertonia persisted (11.24±7.21), appeared after baseline (17.0±10.39), or remained absent (15.79±7.11).")
Progression and clinical worsening of hypertonia (changes in ALSFRS scores at 6 and 12 months from baseline). (A) At 6 months, clinical worsening was more marked in patients in whom hypertonia decreased over the course of the disease (12.33±5.16) (P<.001) than in those in whom spasticity persisted (4.69±4.0), appeared after baseline (11.14±9.81), or remained absent (5.92±3.69). (B) At 12 months, clinical worsening was also more marked in those patients in whom hypertonia disappeared (22.83±9.99) (P<.01) than in those in whom hypertonia persisted (11.24±7.21), appeared after baseline (17.0±10.39), or remained absent (15.79±7.11).
In our study, 26.1% of the patients showed no pyramidal signs at baseline, with a median of 4.5 months from baseline to onset of the first sign. Some pyramidal signs are more frequent than others: hyperreflexia is the most common (69.2%), followed by Babinski sign (49.2%) and spasticity (16.9%). Other authors have reported similar frequencies of Babinski sign (50%) in patients with ALS.5 According to the literature, hyperreflexia and spasticity are frequently difficult to detect or may even be absent in these patients due to weakness and muscle atrophy.6
Furthermore, the resolution of spasticity was associated with more severe clinical deterioration at 6 and 12 months in our study. The resolution of spasticity may be explained by a predominance of signs of lower motor neuron lesions, which have been found to have a greater impact on survival and respiratory function than each pyramidal sign separately.7
We now know that degeneration in ALS affects not only the corticospinal tract and alpha motor neurons, but also the rubrospinal, vestibulospinal, and reticulospinal tracts8; gamma and beta motor neurons9; Renshaw cells; and the proprioceptive pathway.10-12 The complex association between degeneration of the pyramidal system, lower motor neurons, and other tracts, both in the central and in the peripheral nervous systems, explains why pyramidal signs occasionally appear at different frequencies and change as the disease progresses.10 Although pyramidal signs do not change over the course of the disease in most patients, the existence of patterns of exacerbation and of resolution of these signs suggests that there is no direct association between upper and lower motor neuron dysfunction.13,14
ConclusionsOur results underscore the need to develop new neurophysiological15 and imaging tools16 to evaluate pyramidal tract dysfunction, in the same way that electromyography is used to detect lower motor neuron dysfunction. Although detecting upper motor neuron dysfunction has no important therapeutic consequences, it does inform prognosis, due to the ability to establish a more accurate diagnosis, especially in those cases where diagnosis is difficult.
The literature describes multiple neurophysiological and radiological techniques that may be useful for detecting upper motor neuron dysfunction. However, most of these have not been proven to be useful as diagnostic tools for patients with suspected motor neuron disease and no pyramidal signs or symptoms. Only specific transcranial magnetic stimulation protocols (short-interval, paired-pulse stimulation17), triple-pulse transcranial magnetic stimulation,18 and threshold tracking transcranial magnetic stimulation for cortical hyperexcitability15 have enabled the detection of pyramidal tract alterations even at early stages of the disease. However, these techniques are complex and their use in diagnosis is not widespread.
These protocols should be standardised and validated as tools for early diagnosis to complement the existing diagnostic criteria.
Conflicts of interestThe authors have no conflicts of interest to declare.
Please cite this article as: Álvarez N, Díez L, Avellaneda C, Serra M, Rubio MÁ. Relevancia del síndrome piramidal en la esclerosis lateral amiotrófica. Neurología. 2018;33:8–12.
