



Los signos piramidales (hiperreflexia, espasticidad, signo de Babinski) son fundamentales para el diagnóstico de esclerosis lateral amiotrófica (ELA). Sin embargo, no siempre están presentes al comienzo, pueden variar con el tiempo y es controvertido su papel en la evolución. El objetivo del estudio es describir qué signos piramidales están presentes inicialmente y cómo evolucionan en una cohorte de pacientes con ELA, así como su papel pronóstico.
MétodosAnálisis retrospectivo de pacientes recogidos de manera prospectiva, diagnosticados de ELA en nuestro centro, desde 1990 hasta 2015.
ResultadosDel total de 130 pacientes con ELA, 34 (26,1%) no presentaron inicialmente ningún signo piramidal, mientras que 15 (11,5%) presentaban un síndrome piramidal completo. De aquellos pacientes sin piramidalismo inicial, la mediana de aparición de los primeros signos fue de 4,5 meses. El signo de Babinski estaba presente en 64 (49,2%), la hiperreflexia en 90 (69,2%) y en 22 (16,9%) pacientes existía espasticidad. Los signos piramidales tendían a mantenerse inalterados en el tiempo, aunque existe un porcentaje de pacientes en el que aparecen tardíamente o desaparecen con el tiempo.
No se encontró asociación entre supervivencia y la presencia o modificación de signos piramidales, aunque la disminución de la espasticidad se asociaba a mayor deterioro clínico (escala ALSFR) (p<0,001).
ConclusiónUna cuarta parte de pacientes con ELA no presentaron inicialmente ningún signo piramidal y, en algunos casos, estos desaparecen con el tiempo. Esto resalta la necesidad de la inclusión de herramientas para la valoración de la vía piramidal.
Pyramidal signs (hyperreflexia, spasticity, Babinski sign) are essential for the diagnosis of amyotrophic lateral sclerosis (ALS). However, these signs are not always present at onset and may vary over time, besides which their role in disease evolution is controversial. Our goal was to describe which pyramidal signs were present and how they evolved in a cohort of patients with ALS, as well as their role in prognosis.
MethodsRetrospective analysis of prospectively collected patients diagnosed with ALS in our centre from 1990 to 2015.
ResultsOf a total of 130 patients with ALS, 34 (26.1%) patients showed no pyramidal signs at the first visit while 15 (11.5%) had a complete pyramidal syndrome. Of those patients without initial pyramidal signs, mean time of appearance of the first signs was 4.5 months. Babinski sign was positive in 64 (49.2%) patients, hyperreflexia in 90 (69.2%) and 22 (16.9%) patients had spasticity. Pyramidal signs tended to remain unchanged over time, although they seem to appear at later stages or even disappear with time in some patients.
We found no association between survival and the presence of changes to pyramidal signs, although decreased spasticity was associated with greater clinical deterioration (ALSFR scale) (P<.001).
ConclusionA quarter of patients with ALS initially showed no pyramidal signs and in some cases they even disappear over time. These data support the need for tools that assess the pyramidal tract.
La afectación combinada de la vía piramidal yde la segunda motoneurona es el rasgo patológico característico de la esclerosis lateral amiotrófica (ELA)1. Para el diagnóstico se ha establecido que se precisa de la evidencia de afectación de la segunda motoneurona, ya sea clínicamente o mediante un estudio neurofisiológico2, mientras que la demostración de la alteración de la vía piramidal se basa tan solo en el juicio clínico. Sin embargo, los signos piramidales no siempre están presentes en el momento inicial, pueden cambiar con el tiempo, su valoración varía tanto inter- como intraexplorador y se desconoce su papel con relación al pronóstico y evolución de la enfermedad3,4.
El objetivo del estudio es realizar un análisis descriptivo de la presencia de signos piramidales en una cohorte de pacientes con ELA, al inicio y su evolución con el tiempo, así como estudiar su relación con el pronóstico.
MétodosSe realizó un análisis retrospectivo de una base de datos prospectiva, de pacientes diagnosticados de enfermedad de motoneurona en nuestro centro desde 1991 hasta 2015. Se incluyó a aquellos pacientes que cumplían criterios de probable o definida ELA según criterios revisados de El Escorial2 y se recogieron los datos clínicos, epidemiológicos, de situación funcional medida según escala ALSFRS a los 6 y 12 meses y datos sobre la supervivencia (calculada desde inicio de síntomas hasta fallecimiento). Se excluyó a todos los pacientes que presentaban afectación pura de primera (esclerosis lateral primaria) o segunda motoneurona (atrofia muscular progresiva) y a aquellos de los que no se dispusiera de datos sobre su evolución. Se registraron datos de los signos piramidales: tono muscular (aumentado, disminuido, normal), reflejos osteotendinosos (exaltados, presentes, disminuidos/ausentes); reflejo cutáneo plantar (extensor, flexor/indiferente) en la visita inicial y en cada visita sucesiva (media de 3 meses entre visitas) hasta la visita final (última antes del fallecimiento del paciente o antes de la fecha final del estudio). Se consideraba un signo piramidal como presente cuando aparecía en al menos 2 extremidades en el caso de la hipertonía y la hiperreflexia, o al menos en una extremidad si se trataba del signo de Babinski. Si existía algún cambio en un signo piramidal que se confirmaba en al menos 2 visitas sucesivas, se registraba como un cambio. La exploración neurológica fue realizada por 2 neurólogos expertos. Este estudio fue aprobado por el comité ético de nuestro centro (Hospital del Mar, Barcelona).
Análisis estadísticoPara la comparación entre 3 grupos, se usó el test de chi cuadrado para variables categóricas y el de Kruskal-Wallis para variables cuantitativas. En el caso de comparar únicamente 2 grupos, se utilizó el test de la U de Mann Whitney de variables cuantitativas y el test exacto de Fisher en caso de datos categóricos. Se analizó la relación de la supervivencia mediante el método de Kaplan Meier y las curvas obtenidas se compararon usando el test long-rank. Se llevó a cabo un análisis de supervivencia multivariable utilizando el modelo de regresión de Cox, incluyendo las variables que previamente habían mostrado una diferencia estadísticamente significativa (p>0,05) en el método de Kaplan Meier. Para los análisis de datos se usó el software versión 19.0 del SPSS. La fecha final de inclusión de datos fue el 1 de mayo de 2015.
ResultadosDe un total de 256 pacientes con enfermedad de motoneurona, se descartó a 40 pacientes con atrofia muscular progresiva, a 12 con esclerosis lateral primaria y a 74 de los que no se disponía de datos sobre toda su evolución clínica: se obtuvo un total de 130 pacientes diagnosticados de ELA. De estos, 67 (51,5%) eran varones y 63 mujeres (48,5%), con una mediana de edad de 66 años. Del total, 49 pacientes (37,7%) presentaban una forma de inicio bulbar, mientras que 81 (62,3%) comenzaron con una forma espinal. Un total de 112 pacientes (86,1%) habían fallecido en el momento del análisis, con una mediana de supervivencia de 29 meses.
En cuanto a la valoración funcional medida según la escala ALSFRS, en la visita inicial la mediana era de 37 puntos, con un empeoramiento de 5 y 14 puntos, a los 6 y 12 meses respectivamente, respecto el inicio de la enfermedad.
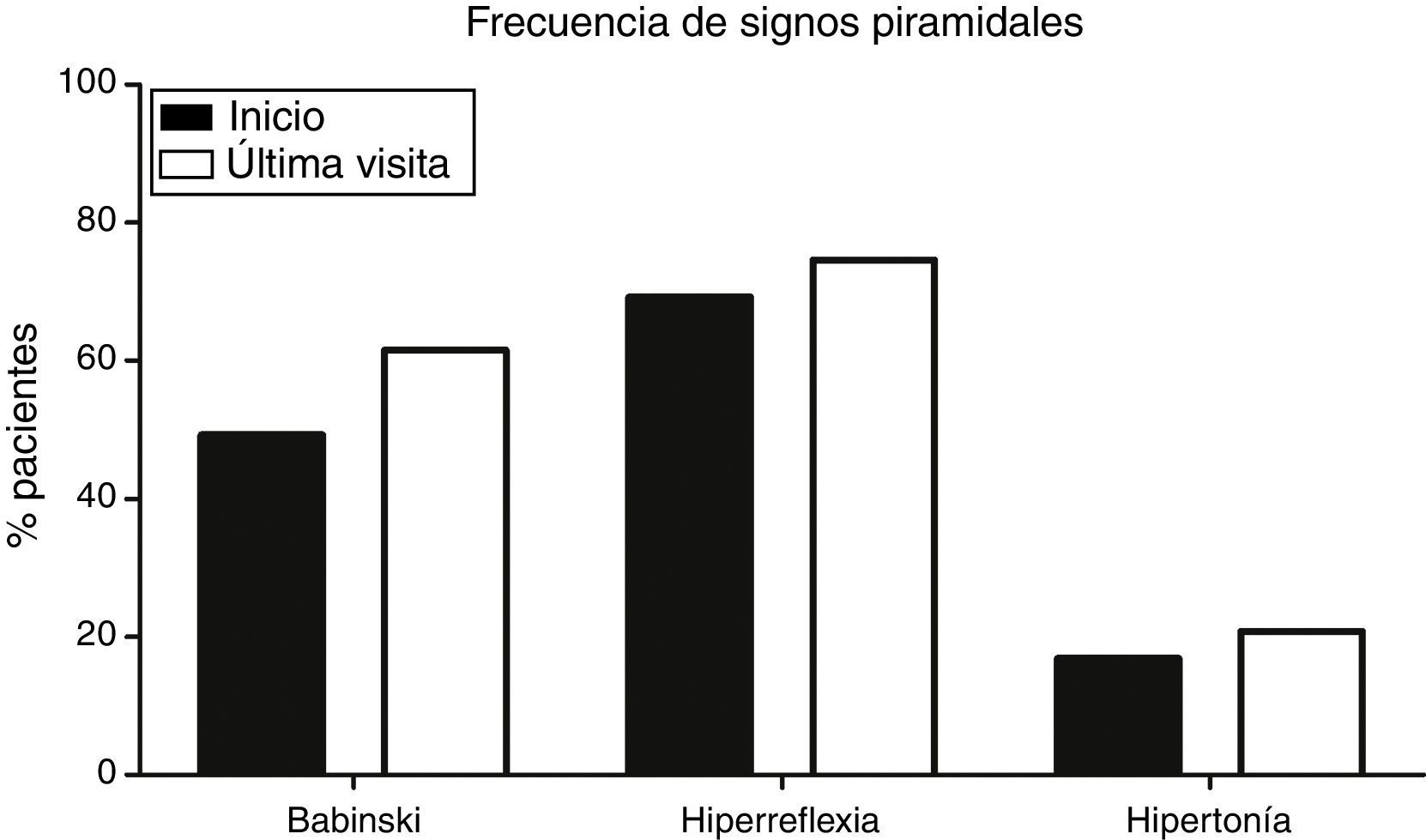
Signos piramidales en la visita inicialDel total de 130 pacientes, 15 (11,5%) presentaban un síndrome piramidal completo en el inicio; es decir, Babinski, hiperreflexia y aumento del tono muscular. Sin embargo, 34 (26,1%) pacientes no presentaron inicialmente ningún signo piramidal, con una mediana de aparición del primer signo de 4,5 meses. Tanto el signo de Babinski, presente en 64 (49,2%) pacientes, como la hiperreflexia, que aparecía en 90 (69,2%), eran más frecuentes (p<0,001) comparados con la frecuencia de espasticidad inicial (p<0,001), que tan solo estaba presente en 22 (16,9%) pacientes (fig. 1).
 y la hiperreflexia (69,2% al inicio; 74,6% final) eran significativamente más frecuentes (p<0,001) que la hipertonía, tanto al inicio (16,9%) como en la visita final (20,8%).")
Frecuencia de los signos piramidales en la visita inicial y la última visita. El signo de Babinski (49,2% al inicio; 61,5% final) y la hiperreflexia (69,2% al inicio; 74,6% final) eran significativamente más frecuentes (p<0,001) que la hipertonía, tanto al inicio (16,9%) como en la visita final (20,8%).
No se encontraron diferencias estadísticamente significativas en cuanto a sexo, forma de inicio (espinal o bulbar) y supervivencia entre aquellos pacientes que presentaban o no cualquiera de los signos piramidales, así como tampoco en aquellos con un síndrome piramidal completo desde el inicio.
Evolución de signos piramidalesTodos los pacientes, durante el transcurso de su enfermedad, acaban presentando signos piramidales, condición indispensable para el diagnóstico de ELA. Sin embargo, con el tiempo, algunos signos que inicialmente están presentes pueden desaparecer: en nuestra cohorte un total de 12 (9,2%) pacientes no presentan ningún signo piramidal en la visita final. Solo 24 (18,5%) pacientes presentaban en la visita final un síndrome piramidal completo. De nuevo, tanto el signo de Babinski (80 pacientes; 61,5%) como la hiperreflexia (97 pacientes; 74,6%) eran más frecuentes en la visita final (p<0,001) que la hipertonía (27 pacientes; 20,8%) (fig. 1).
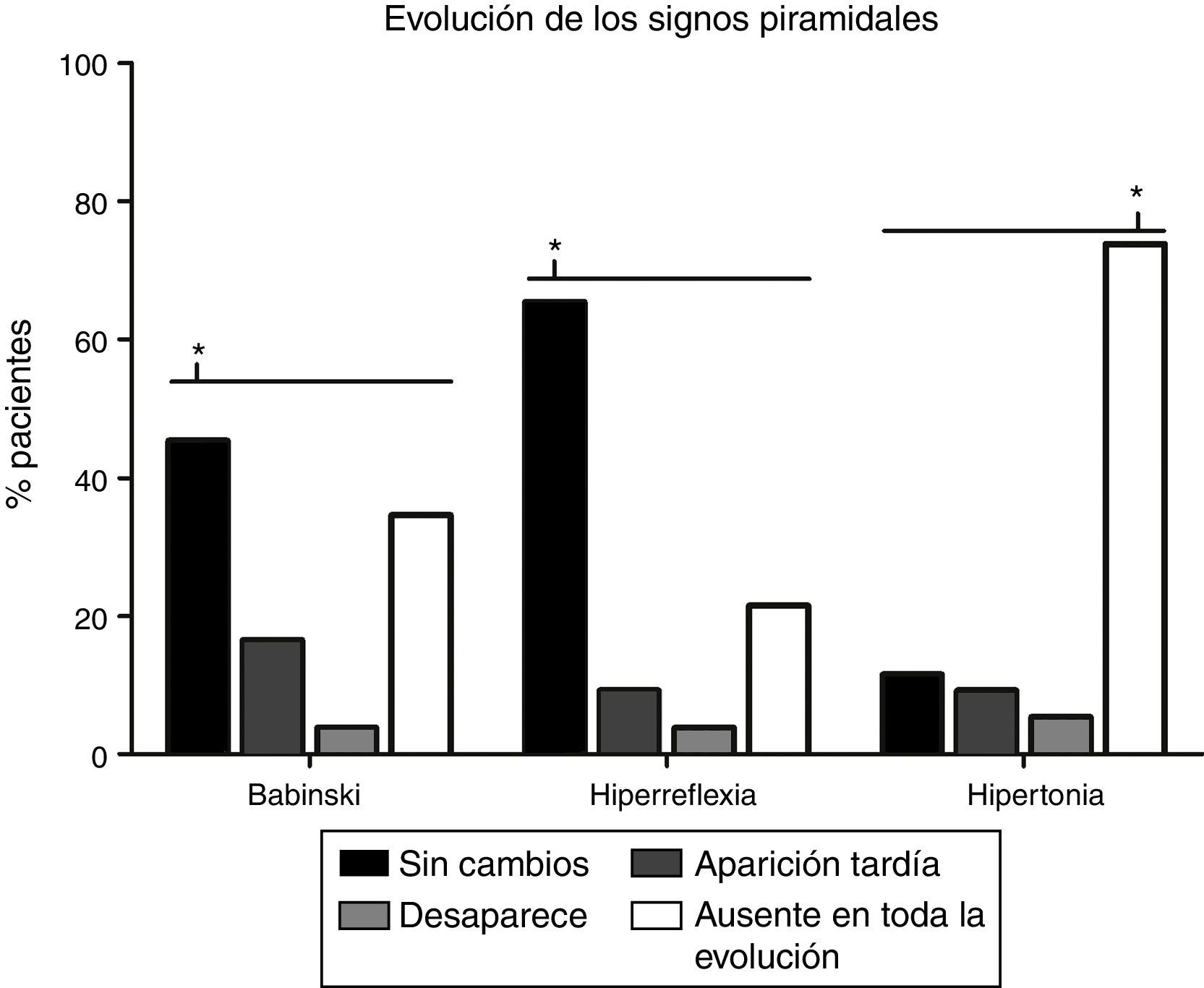
La evolución de los signos piramidales está descrita en la figura 2. Tanto para la hiperreflexia (65,4%) como para el signo de Babinski (45,4%), la evolución más frecuente era que se mantuviesen sin cambios desde la visita inicial (p<0,0001). En cuanto al tono muscular, era más frecuente (p<0,0001) la ausencia de espasticidad (73,8%) en todo el transcurso de la enfermedad.
 y el signo de Babinski (45,4%) es que se mantengan sin cambios desde el inicio (p<0,0001), mientras que, con relación al tono muscular, la ausencia de espasticidad en todo el transcurso de la enfermedad (73,8%) es la evolución más frecuente (p<0,0001). El signo de Babinski aparecía posteriormente a la visita inicial en 21 pacientes (16,5%), desaparecía en 5 (3,8%) y en 45 pacientes (34,6%) nunca estuvo presente. La hiperreflexia aparecía tardíamente en 12 pacientes (9,2%), desaparecía en 5 (3,8%) y estaba ausente en toda la evolución en 28 pacientes (21,5%). Aparecía espasticidad cuando inicialmente no existía en 12 pacientes (9,2%), disminuía el tono muscular en 7 (5,4%), mientras que, en 15 (11,5%) casos, la hipertonía permanecía inalterada.* p<0,0001.")
Análisis de los cambios en los signos piramidales con la evolución de la enfermedad. La evolución más frecuente en el caso de la hiperreflexia (65,4%) y el signo de Babinski (45,4%) es que se mantengan sin cambios desde el inicio (p<0,0001), mientras que, con relación al tono muscular, la ausencia de espasticidad en todo el transcurso de la enfermedad (73,8%) es la evolución más frecuente (p<0,0001). El signo de Babinski aparecía posteriormente a la visita inicial en 21 pacientes (16,5%), desaparecía en 5 (3,8%) y en 45 pacientes (34,6%) nunca estuvo presente. La hiperreflexia aparecía tardíamente en 12 pacientes (9,2%), desaparecía en 5 (3,8%) y estaba ausente en toda la evolución en 28 pacientes (21,5%). Aparecía espasticidad cuando inicialmente no existía en 12 pacientes (9,2%), disminuía el tono muscular en 7 (5,4%), mientras que, en 15 (11,5%) casos, la hipertonía permanecía inalterada.* p<0,0001.
No se observaron diferencias en cuanto a sexo, edad de inicio, forma de inicio (espinal o bulbar), ni supervivencia entre aquellos pacientes en los que los signos piramidales se modificaban y aquellos en los que permanecían estables.
Signos piramidales y pronósticoNo se encontraron diferencias estadísticamente significativas al analizar el tiempo de supervivencia y la presencia o ausencia de cualquier signo piramidal en el inicio, así como sus cambios en el tiempo. Tampoco se encontró una relación entre el tiempo de supervivencia y la existencia de un síndrome piramidal completo o con la ausencia total de signos piramidales en la visita inicial.
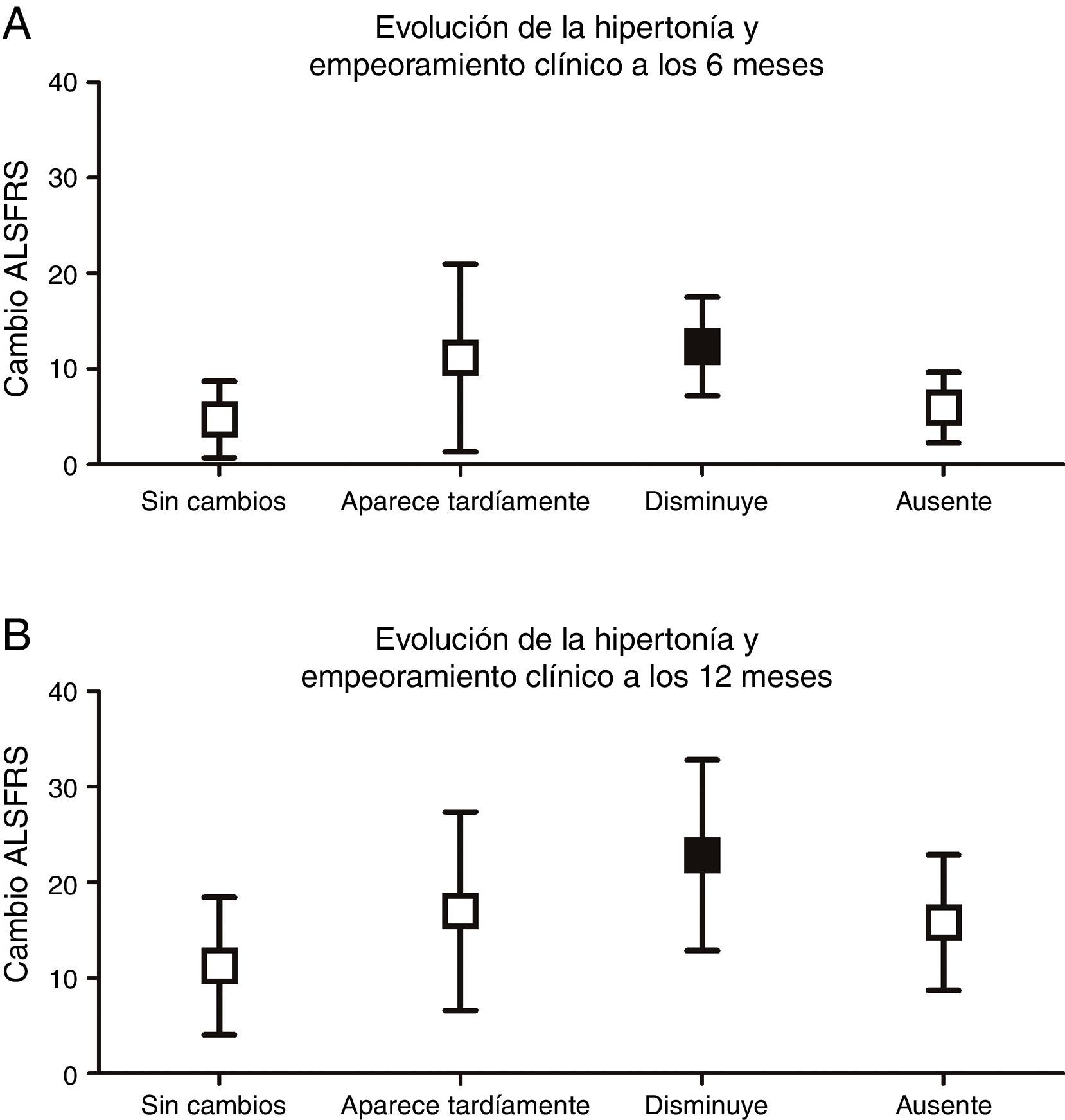
Se analizó si las diferentes progresiones que podía tomar cada signo piramidal se asociaban a un mayor deterioro funcional. Tan solo la desaparición de la espasticidad se asociaba a un mayor deterioro clínico a los 6 (p<0,001) y 12 meses (p<0,01), valorada según la variación de la escala ALSFRS respecto a la visita inicial (fig. 3).
. A) A los 6 meses, mayor empeoramiento en el grupo de la disminución de la hipertonicidad (12,33±5,16) (p<0,001), comparado con el grupo en el que se mantiene la espasticidad (4,69±4,0), aparece tardíamente (11,14±9,81) o está ausente (5,92±3,69). B) A los 12 meses, también hay mayor empeoramiento en el grupo en el que la hipertonía desaparece (22,83±9,99) (p<0,01) comparado con el que se mantiene sin cambios (11,24±7,21), aparece tardíamente (17,0±10,39) o está ausente durante toda la evolución (15,79±7,11).")
Evolución de la hipertonicidad y empeoramiento clínico (variación de la escala ALSFRS a los 6 y a los 12 meses respecto el inicio). A) A los 6 meses, mayor empeoramiento en el grupo de la disminución de la hipertonicidad (12,33±5,16) (p<0,001), comparado con el grupo en el que se mantiene la espasticidad (4,69±4,0), aparece tardíamente (11,14±9,81) o está ausente (5,92±3,69). B) A los 12 meses, también hay mayor empeoramiento en el grupo en el que la hipertonía desaparece (22,83±9,99) (p<0,01) comparado con el que se mantiene sin cambios (11,24±7,21), aparece tardíamente (17,0±10,39) o está ausente durante toda la evolución (15,79±7,11).
En nuestro estudio, hasta un 26,1% de los pacientes no presentaba ningún signo piramidal en la visita inicial, con una mediana de 4,5 meses hasta la aparición del primer signo. Existen unos signos que están presentes con mayor frecuencia que otros: la hiperreflexia en primer lugar (69,2%), el signo de Babinski en segundo (49,2%) y, por último, la espasticidad (16,9%). Otros autores han reportado una proporción similar de pacientes con signo de Babinski (50%) en pacientes con ELA5. También se ha descrito que, a menudo, tanto la hiperreflexia como la espasticidad pueden ser difíciles de detectar o estar ausentes en estos pacientes debido a la debilidad y a la atrofia muscular6.
Además, hemos encontrado que la desaparición de la espasticidad se asocia a un mayor deterioro clínico a los 6 y 12 meses. La desaparición de espasticidad se explicaría por un predominio de los signos de afectación de la segunda motoneurona, los cuales se ha visto que tienen una mayor repercusión, no solo en la supervivencia, sino también en la función respiratoria, si se comparan con los signos piramidales aislados7.
Actualmente se conoce que existe una degeneración que va más allá de la vía corticoespinal y la motoneurona alfa: tracto rubroespinal, vestibuloespinal, reticuloespinal8, motoneuronas beta y gamma9, interneuronas de Renshaw y la vía propioceptiva10–12. La compleja relación entre la degeneración del sistema piramidal, la motoneurona inferior y demás vías, tanto del sistema nervioso central como del periférico, es la responsable de que, en ocasiones, los distintos signos piramidales se presenten en diferente frecuencia y puedan variar con la propia evolución de la enfermedad10. Si bien, en la mayoría de los pacientes, los signos piramidales no se modificaban con el tiempo, la existencia de un patrón de exaltación de los signos piramidales y otro de desaparición indica que no parece existir una relación directa entre la afectación de la primera y la segunda motoneuronas13,14.
ConclusionesTodos estos datos resaltan la necesidad de otras herramientas para la valoración de la vía piramidal, ya sea mediante técnicas neurofisiológicas15, ya sea mediante pruebas de imagen16, del mismo modo que se utiliza el electromiograma para la demostración de la implicación de la segunda motoneurona. Aunque hoy día carece de una implicación terapéutica relevante, sí que resulta importante su evaluación para establecer un pronóstico ante un diagnóstico más preciso, sobre todo en aquellas formas de difícil diagnóstico en una primera instancia.
Se han descrito múltiples técnicas, tanto neurofisiológicas como radiológicas, para poner de manifiesto la afectación de la vía piramidal. Sin embargo, la mayoría no han demostrado su utilidad como herramienta diagnóstica en pacientes con sospecha de enfermedad de motoneurona y ausencia de signos y síntomas piramidales. Tan solo el uso de un determinado protocolo de estimulación magnética transcraneal (short-interval, paired-pulse stimulation17), de la estimulación magnética transcraneal con triple estímulo18 y de la hiperexcitabilidad cortical demostrada mediante threshold tracking15 han permitido detectar alteraciones de la vía piramidal, ya incluso en estadios iniciales. No obstante, todas estas técnicas son complejas y no se utilizan con regularidad en todos los centros, con lo que su uso generalizado como ayuda diagnóstica está limitado.
Por ello, es necesario la homogeneización de estos protocolos entre centros, así como su validación como herramienta de diagnóstico precoz de cara a valorar su complementariedad con los criterios diagnósticos actuales.
Conflicto de interesesNinguno de los autores presenta conflicto de interés con relación a este artículo.

