




Prion diseases are neurodegenerative disorders resulting from the accumulation of a misfolded isoform of the cellular prion protein (PrPC). They can occur as acquired, sporadic, or hereditary forms. Although prion diseases show a wide range of phenotypic variations, pathological features and clinical evolution, they are all characterised by a common unfavourable course and a fatal outcome.
Review summarySome variants, such as kuru, have practically disappeared, while others, for example the variant Creutzfeldt–Jakob disease (vCJD) or those attributable to iatrogenic causes, are still in force and pose a challenge to current medicine. There are no definitive pre-mortem diagnostic tests, except for vCJD, where a tonsil biopsy detects 100% of the cases. For this reason, diagnostic criteria dependent on statistical probability have had to be created. These require complementary examinations, such as an electroencephalogram (EEG) or the detection of 14-3-3 protein in cerebrospinal fluid (CSF). Only the pulvinar sign in magnetic resonance imaging (MRI) has been included as a vCJD diagnostic criterion. The present review discusses neuroimaging findings for each type of prion disease in patients with a definitive histopathological diagnosis.
ConclusionsThe aim is to define the usefulness of these complementary examinations as a tool for the diagnosis of this family of neurodegenerative diseases.
Las prionopatías son un conjunto de enfermedades neurodegenerativas producidas por el acúmulo de una isoforma anormal de la proteína priónica celular (PrPc). Se clasifican en adquiridas, hereditarias y esporádicas. Aunque son muchas las características clínicas, evolutivas y anatomopatológicas que las diferencian, todas ellas tienen en común un curso desfavorable y un pronóstico fatal.
DesarrolloSi bien algunos tipos como el kuru, prácticamente han desaparecido, otras formas como la variante de la enfermedad de Creutzfeldt-Jakob (vECJ) o la iatrogénica siguen vigentes y suponen un reto en la medicina actual. El diagnóstico de certeza se realiza postmortem, salvo en el caso de la vECJ en la que la biopsia amigdalar puede detectar el 100% de los casos. Ello ha supuesto que se definan criterios diagnósticos en función de una probabilidad estadística, que precisa la realización de exámenes complementarios tales como el estudio electroencefalográfico y la detección de la proteína 14-3-3 en el líquido cefalorraquídeo (LCR). Solamente la vECJ ha llegado a incluir en los criterios diagnósticos de la OMS el «signo del pulvinar» en resonancia magnética cerebral (RM). En este artículo se revisan los hallazgos de neuroimagen que han sido descritos para cada tipo de prionopatía en pacientes con un diagnóstico de certeza.
ConclusionesLa finalidad es definir la utilidad de estas pruebas complementarias como una herramienta de apoyo para el diagnóstico de este conjunto de enfermedades neurodegenerativas.
Prion diseases belong to a group of neurodegenerative diseases that are caused by post-transcriptional conversion of the cellular prion protein (PrPC) into an abnormal isoform named PrPSc. This misfolded protein has biochemical and biophysical properties allowing it to act as an infectious agent in the absence of nucleic acids.1
PrPC is a cell membrane glycoprotein that is encoded by the PRNP gene on chromosome 20. This gene is polymorphic at codon 129, resulting in either valine (V) or methionine (M). Methionine homozygosity is a risk factor for developing prion diseases. PrPC is synthesised in the cytoplasm and passes through the endoplasmic reticulum and Golgi apparatus before reaching the plasma membrane, where its main location is the extracellular matrix.2 The mature form of this protein is composed of 209 amino acids with a disulphide bond.3
PrPC expression begins in the earliest stages of embryogenesis. In adults, the protein is present in neurons of the brain and spinal cord, and to a lesser extent in glial cells in the central nervous system (CNS) and different cell types found in musculoskeletal, cardiac, and lymphoreticular tissue.4 The function of PrPC is unknown, although different hypotheses state that it may participate in neural stem cell differentiation and neurogenesis,5 long-term renewal of haematopoietic stem cells,6 lymphocyte activation,7 neuroprotection from apoptosis and oxidative stress,8 transmembrane signalling,9 synapse formation and maintenance,10 copper ion binding,11 and cell–extracellular matrix adhesion. Deficiency of this protein does not elicit detectable disease12; in contrast, PrPC must be expressed in order for prion disease to appear in the host.13 Elevated levels of PrPSc alone do not cause prion diseases to develop. Changes in PrPC also contribute to the process, and there are several hypotheses explaining how transformation of that protein may cause neurodegeneration.14
The characteristics common to all prion diseases are damage limited to the CNS, a lengthy incubation period, and a progressive, invariably lethal course of the disease.3 One of the most common clinical manifestations of these diseases is onset of dementia. All prion diseases also share neuropathological characteristics that include neuronal loss, reactive gliosis, amyloid plaques of PrPC, and the large vacuoles that cause the brain's typical spongy appearance. Historically, prion diseases have been categorised as acquired, hereditary, or sporadic.3 The following section describes the clinical manifestations and main neuroimaging findings in different prion diseases.
DevelopmentAcquired formsKuruKuru is a prion disease of great historical interest that was discovered in the 1950s. The disease reached epidemic status among certain groups in Papua New Guinea, and its transmission is related to cannibalistic practices. It affected women and children almost exclusively, since men did not participate in funeral preparations; also, when practising endocannibalism, they tended to consume muscle tissue rather than brain or viscera. In the local language, ‘kuru’ means ‘to tremble or shake’, which is one of the essential clinical characteristics of the disease, together with progressive cerebellar ataxia, choreoathetosis, and abnormal eye movements. Clinical development of these symptoms is preceded by a prodromal phase with headache and muscle and joint soreness. Kuru has a long incubation period and its incidence rate has decreased since the cessation of cannibalism.15 Genetic studies revealed that individuals who were exposed to the disease but did not develop it were heterozygous for the codon 120 polymorphism in the PRNP gene. Another more recent study indicates that the 127V polymorphism is also a factor protecting individuals from developing kuru.16 No neuroimaging studies of this disease are available.
Iatrogenic Creutzfeldt–Jakob diseaseIatrogenic Creutzfeldt–Jakob disease (CJD) develops after person-to-person transmission of the infection as the result of any of a list of medical or surgical procedures. The first case was described in 1974 in a patient who received a corneal transplant from a donor and later died of CJD.17 Since then, cases of CJD have been described after use of contaminated equipment in neurosurgical procedures, intracerebral electrode placement, pituitary hormone administration, dura mater or corneal transplants, and more recently, blood transfusions.18
Diagnosis is based on the presence of typical, compatible symptoms including dementia, ataxia, pyramidal and extrapyramidal signs, myoclonia, and akinetic mutism,19 plus prior exposure to the disease. Elevated 14-3-3 protein can be found in cerebrospinal fluid (CSF) in 77% of all cases.20 Electroencephalographic findings are similar to those in sporadic CJD and include periodic spike–wave complexes that are sometimes more prominent at the inoculation site.21 Histological studies show the same typical findings mentioned previously, although researchers have also described diffuse plaques of PrP in a variant of the form caused by dura mater transplant. The course of this disease variant is insidious and predominated by ataxia and dementia. Electroencephalographic changes typically do not appear before the final stages of CJD.19
Both clinical symptoms and the incubation period vary depending on the route of inoculation. In the case of intracerebral inoculation, or inoculation near the CNS, incubation lasts only months and dementia is the predominant symptom. Peripheral inoculation gives rise to a predominantly ataxic syndrome reminiscent of kuru, with an incubation period of up to 31 years. Most patients are MM homozygous at codon 129, which stresses the importance of this genetic trait as a marker for susceptibility.
Meissner et al.19 used magnetic resonance imaging (MRI) to study a series of 10 patients who developed CJD following a dura mater transplant. They observed cortical hyperintensities in these patients, affecting (in order of frequency) the cingulate gyrus and the frontal, temporal, and parietal lobes in sequences taken with DWI (diffusion-weighted imaging) and FLAIR (fluid-attenuated inversion recovery). Changes in the occipital cortex are less common, but the basal ganglia and the thalamus will also be affected in a high number of patients. However, the pulvinar or ‘hockey-stick’ sign that characterises variant CJD does not appear in the thalamus.
Similar findings have been described in cases of iatrogenic CJD due to a corneal transplant. DWI and FLAIR sequences reveal extensive diffusion-restricted lesions in the parieto-occipital cortex and frontotemporal lobes, together with bilateral putaminal hyperintensities and unilateral intensities in the head of the caudate nucleus.22
The same changes have been described in patients whose CJD was transmitted through human growth hormone therapy. One interesting feature in these cases is cerebellar involvement, which is uncommon in other types of CJD.23 This finding, in conjunction with compatible symptoms, may support diagnosis of this disease.
Spectroscopy studies have not yet been able to establish a standard pattern for the disease, although they do show that areas with a decreased peak for N-acetyl aspartate (NAA) correspond to areas of progressive neuronal death.22,24
New variant of Creutzfeldt–Jakob disease‘Mad cow disease’ or vCJD was first described in 1996. It is caused by the same agent that causes bovine spongiform encephalopathy.25 Variant CJD affects younger patients than sporadic CJD does, and its median survival time is 14 months. A recent article describes a familial case of the disease.26 The initial clinical course of vCJD is marked by psychiatric symptoms, including changes in behaviour, mood swings, irritability, and social withdrawal. Subsequently, half of all patients present sensory symptoms manifesting as dysaesthesia in the extremities and facial region; facial pain is typical. As the disease progresses, patients experience ataxia associated with involuntary movements reminiscent of those in kuru which may be choreic, dystonic, or myoclonic. Progressive cognitive decline will eventually appear and progress to akinetic mutism.27 Electroencephalogram (EEG) studies show non-specific abnormalities and in 50% to 60% of all cases, 14-3-3 protein can be detected with specificity greater than 94%. Noteworthy findings in anatomical pathology studies include the presence of ‘florid plaques’, PrPSc plaques surrounded by areas of spongiform change.28 As in other prion diseases, a neuropathological examination determines the definitive diagnosis of vCJD. In this variant, however, histological study of the tonsils will detect PrPSc in 100% of the cases. Living subjects can therefore receive a firm diagnosis.
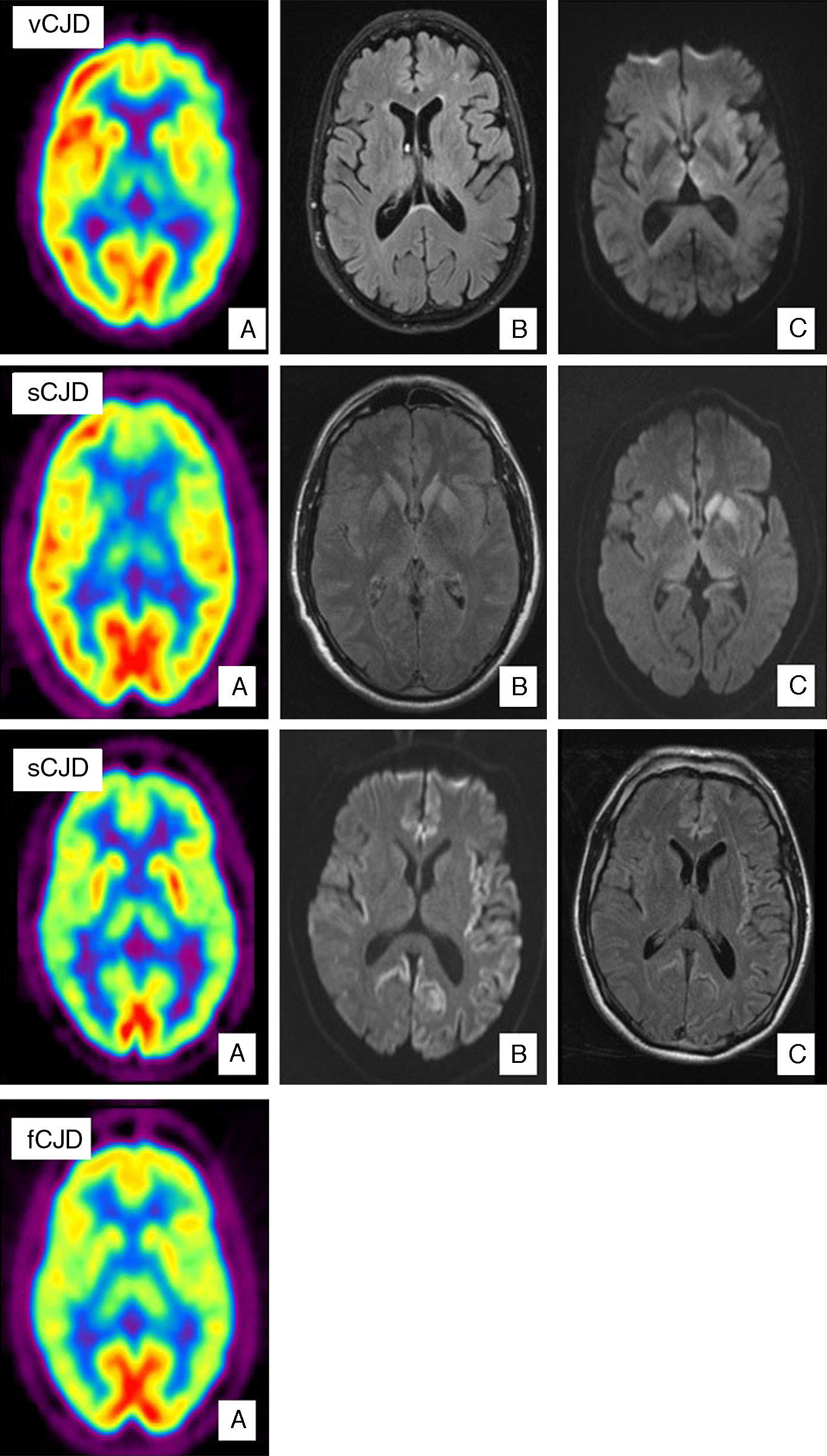
Variant CJD is a clear example of how imaging tests have become increasingly important. Proof of this tendency is that the pulvinar sign is included in the WHO diagnostic criteria for the disease (Table 1). The pulvinar sign refers to a pattern of symmetrical hyperintensities in both pulvinar thalamic nuclei with respect to the cerebral cortex and the anterior part of the putamen as observed by MRI. It can be detected using T2-weighted, proton density-weighted, FLAIR, and DWI sequences. Its diagnostic sensitivity for vCJD ranges from 78% to 90% and its specificity approaches 100%.29 Another characteristic finding is called the ‘hockey stick sign’; here, the pulvinar sign appears along with hyperintensities in the dorsomedial thalamic nuclei.29 This occurs in 56% of all cases (Fig. 1). These MRI findings may be the result of astrocytosis in the pulvinar nuclei.29,30 In addition to gliosis, neuronal loss and neuropil vacuolisation may also occur at this location and in the dorsomedial nuclei. Nevertheless, spongiform changes are more prominent in the caudate nucleus and putamen. These changes have not been correlated with any specific MRI patterns. A recent study describes the ‘hot-cross bun’ sign in a case of vCJD. This sign refers to a cross-shaped pontine hyperintensity characteristic of multiple system atrophy.31 Likewise, hyperintensity in periaqueductal grey matter has been described in a small number of cases. Differential diagnosis of patients with MRI signal changes in the thalamus must consider sporadic prion diseases. MRI in cases of sporadic prion disease will also reveal thalamic abnormalities without the pulvinar sign; in addition, thalamic hyperintensity tends to be less pronounced, and more asymmetrical, than that in the head of the caudate nucleus.32 Candidate conditions should also include intracranial hypertension, cat scratch disease,33 Alpers disease,34 paraneoplastic syndromes,35 and post-infectious encephalitis.36
CJD diagnostic criteria.
| I: |
| A. Progressive neuropsychiatric changes |
| B. Course of the disease >6 months |
| C. No alternative diagnosis explaining the symptoms |
| D. No history of possible iatrogenic exposure |
| E. No evidence suggesting that CJD could be familial |
| II: |
| A. Early-onset psychiatric symptoms |
| B. Painful and persistent sensory symptoms |
| C. Ataxia |
| D. Myoclonia, chorea, or dystonia |
| E. Dementia |
| III: |
| A. EEG does not show typical signs of the sporadic form (or EEG not performed) |
| B. MRI shows bilateral, symmetrical hyperintensities in the pulvinar |
| IV: |
| A. Positive tongue biopsy |
| Definitive: IA and neuropathological confirmation for vCJD |
| Probable: I and 4/5 II and IIIA and IIIB; I and IVA |
| Possible: I and 4/5 of type II and IIIA |
 PET-FDG shows bilateral thalamic and left hemisphere hypometabolism. (B) FLAIR sequence showing no relevant changes. (C) Bilateral signal increase in the dorsomedial nucleus of the thalamus in diffusion sequences. Case of sCJD. (A) PET-FDG showing hypometabolic areas in the caudate nucleus, thalamus, and anterior region of the putamen. Hypometabolism in the frontal pole and dorsomedial area of the frontal lobe, predominantly on the left side. (B) Bilateral hyperintensities in the basal ganglia and the genu of the left internal capsule in FLAIR sequences. (C) Similar findings in diffusion sequences also showing hyperintensity in the thalamus and cingulate cortex. Probable case of sCJD (MM2 subtype). (A) PET-FDG scan showing generalised reduction in activity in the cortex and both thalamic regions. (B) Bihemispheric, predominantly left-sided cortical hyperintensity in FLAIR sequences. (C) Similar findings in diffusion sequences. Case of fCJD. (A) PET-FDG scan showing predominantly right-sided areas of hypometabolism in the thalamic and basal ganglion regions and bilateral impairment of the caudate nucleus.")
Case of vCJD. (A) PET-FDG shows bilateral thalamic and left hemisphere hypometabolism. (B) FLAIR sequence showing no relevant changes. (C) Bilateral signal increase in the dorsomedial nucleus of the thalamus in diffusion sequences. Case of sCJD. (A) PET-FDG showing hypometabolic areas in the caudate nucleus, thalamus, and anterior region of the putamen. Hypometabolism in the frontal pole and dorsomedial area of the frontal lobe, predominantly on the left side. (B) Bilateral hyperintensities in the basal ganglia and the genu of the left internal capsule in FLAIR sequences. (C) Similar findings in diffusion sequences also showing hyperintensity in the thalamus and cingulate cortex. Probable case of sCJD (MM2 subtype). (A) PET-FDG scan showing generalised reduction in activity in the cortex and both thalamic regions. (B) Bihemispheric, predominantly left-sided cortical hyperintensity in FLAIR sequences. (C) Similar findings in diffusion sequences. Case of fCJD. (A) PET-FDG scan showing predominantly right-sided areas of hypometabolism in the thalamic and basal ganglion regions and bilateral impairment of the caudate nucleus.
There are very few functional neuroimaging studies in patients with vCJD (Fig. 1).26 Researchers using single photon emission computed tomography (SPECT) with 99mTc-hexamethylpropylene-amine oxime (99mTc-HMPAO) have observed non-specific alterations including diffuse cortical hypoperfusion or more localised changes such as those in the left temporoparietal region.37 MRI studies with spectroscopy in these patients point to increased levels of myo-inositol (Mi) and decreased levels NAA in the thalamus, and to a lesser extent, in the caudate nucleus.38
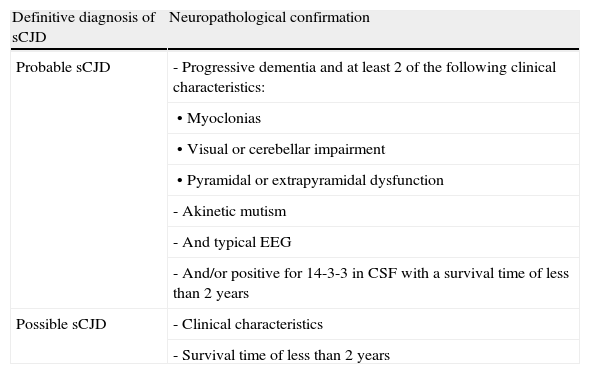
Sporadic formsSporadic Creutzfeldt–Jakob disease (sCJD)This form of CJD accounts for 85% of all cases of human prion disease. The incidence rate is the highest in subjects in their seventh decade. Its prodromal symptoms are non-specific and include weight loss, sleep disorders, behavioural disorders or changes in personality, and vaguely described visual changes. Gait and language are also affected during early stages. These changes are followed by rapidly progressing dementia that may be accompanied by cerebellar ataxia and myoclonia. The last stage of the disease is marked by akinetic mutism with myoclonia and the death of the patient after a median survival time of 6 months. There is no specific diagnostic test for this disease and therefore probability criteria are used (Table 2). A definitive diagnosis can only be confirmed by neuropathological findings.
CJD diagnostic criteria.
| Definitive diagnosis of sCJD | Neuropathological confirmation |
| Probable sCJD | - Progressive dementia and at least 2 of the following clinical characteristics: |
| • Myoclonias | |
| • Visual or cerebellar impairment | |
| • Pyramidal or extrapyramidal dysfunction | |
| - Akinetic mutism | |
| - And typical EEG | |
| - And/or positive for 14-3-3 in CSF with a survival time of less than 2 years | |
| Possible sCJD | - Clinical characteristics |
| - Survival time of less than 2 years |
The first MRI studies performed in patients with sCJD showed symmetrical hyperintensities in the basal ganglia (caudate nucleus and putamen) in T2-weighted and PD-weighted sequences. This finding has a diagnostic sensitivity of 67% and a specificity of 93%.39 Later studies demonstrated the superior results delivered by FLAIR sequences, especially DWI sequences (Fig. 1).39–43 DWI has a higher detection capacity for detecting the disease44 than T2-weighted, PD-weighted or FLAIR sequences. It also has a higher detection capacity than EEG (50%–78%) or CSF assays of 14-3-3 proteins (84%–94%) or neuron-specific enolase (NSE) (73%).45,46 Combined use of FLAIR and DWI delivers an MRI study with a diagnostic sensitivity of 91% and a specificity of 95%.40 Lodi et al.43 recently suggested that current MRI protocols for diagnosing sCJD should also include a study of the thalamus using proton MR spectroscopy (1H NMR). The same authors indicate that using the NAA/Cr ratio in the thalamus in the 1H MRS sequence (cut-off point ≤1.21), plus hyperintensity in the striatum/cortex in DWI sequences, assigns the correct diagnosis to 93% of patients with sCJD.
Two basic patterns in DWI sequences have been defined in patients with sCJD. The first, appearing in two-thirds of all patients, is characterised by hyperintensities in cortical regions and subcortical structures, fundamentally the striatum and medial and/or posterior thalamus. The second is characterised by hyperintensities in cortical areas only. These patterns are believed to have prognostic implications, with a longer median survival time in patients with lesions limited to the cortex.41 No signal alterations are found in 7% of the patients with sCJD, and hyperintensities are limited to the basal ganglia in 2%.
In the group with hyperintensities limited to the cortex in the DWI sequence, 89% present impairment in at least 3 cortical areas that frequently include the parietal and frontal lobes (78%). FLAIR sequences show less extensive cortical impairment; however, FLAIR is more sensitive for detecting hyperintensities in the hippocampus. Both sequences clearly show lack of impairment in the primary somatosensory and visual areas.40,41
Zerr et al.47 recently proposed creating new diagnostic criteria for sCJD that would deliver a sensitivity of 98%. In addition to considering EEG findings and the 14-3-3 protein assay in CSF, these criteria include FLAIR or DWI detection of a hyperintensity in the caudate nucleus or putamen or at least 2 cortical regions.
Possible causes of false positives include encephalitis,48–50 hypoxia,51 epilepsy,52 carbon monoxide poisoning,53 hypoglycaemia, Wilson disease,54 mitochondrial disorders,55 reversible posterior leukoencephalopathy,56 Huntington disease, and other entities that progress with cognitive impairment and which must be ruled out by differential diagnosis.57
Abnormalities in diffusion-weighted MRI studies correspond to spongiform changes that occur because the presence of small vacuoles results in restricted diffusion of free water.58 The fact that vacuoles constitute the key neuropathological trait in the disease's early stages explains why DWI sequences are the most sensitive for diagnosing sCJD. As the disease progresses, gliotic changes become more prominent and easy to detect using T2-weighted and FLAIR sequences.
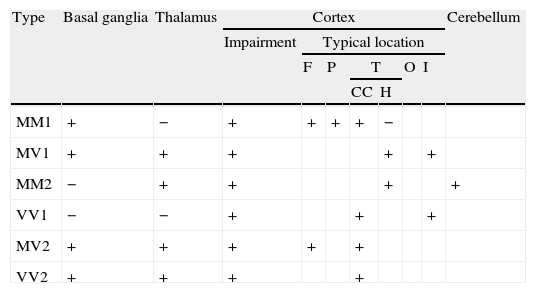
The combination of the genotype at codon 129, and the protein's molecular type according to its behaviour following partial digestion with proteinase K (types 1 and 2), yields 6 molecular subtypes of the disease with different neuropathological and clinical profiles and distinct neuroimaging alterations. MM1 and MV1 are the most common and account for 75% of all sporadic prion diseases.59 Generally speaking, these are rapidly evolving forms that typically present with cognitive deterioration and cortical visual impairment (Heidenhain variant).60 Pseudoperiodic complexes are present in EEG findings and the CSF tests positive for 14-3-3 protein in more than 96% of all cases.45,61 Cortical variants MM2 and VV1 are characterised by early age at onset. Duration of these subtypes is longer; the disease progresses to dementia relatively slowly with early-onset spatial disorientation, aphasia, or apraxia.62 Increased levels of 14-3-3 protein in CSF tend to be present in these subtypes, although EEG studies do not show typical changes. The MV2 subtype progresses with dementia, ataxia, and akinetic rigid syndrome. Exceptionally, myoclonia may appear in final stages. Testing does not detect 14-3-3 protein level alterations in these patients and EEG results are either normal or show non-specific results, so neuroimaging tests may be particularly useful.63 The main initial symptom among patients with the VV2 subtype is cerebellar ataxia, although dementia has also been described as an early sign in a sizeable percentage of these patients. Patients with VV2 occasionally show typical EEG alterations; the most useful diagnostic procedure is assaying 14-3-3 protein in the CSF.45
The next section describes the most typical FLAIR and DWI neuroimaging findings in sCJD according to its different molecular subtypes (Table 3).44
- •
Cortical and basal ganglia impairment:
- ∘
MM1: hyperintensities are often present in basal ganglia.57 Half of all patients with MM1 display extensive cortical hyperintensity that mainly affects the frontal and parietal lobes and cingulate gyrus. Lack of hyperintensities in the hippocampus and thalamus is a typical feature.
- ∘
MV1: hyperintensities are often found in the basal ganglia and cortex and frequently include the insular cortex and hippocampus.57 In contrast with MM1, with which it shares a phenotype, MV1 more commonly affects the cortex and thalamus.
- ∘
- •
Predominantly cortical impairment:
- ∘
MM2 (Fig. 1): Researchers have described extensive cortical impairment that includes the temporal lobes. Hyperintensities in the hippocampus and cerebellum are typical in MM2, and this finding distinguishes it from MM1. Changes in basal ganglia are limited, meaning that lack of hyperintensity at this location supports a diagnosis of MM2. Cortical, thalamocortical and thalamic variants have been described within this subtype.64 The thalamic variant of MM2 is also known as sporadic fatal insomnia.65
Sporadic fatal insomnia was initially described as another clinical entity in 1999.66 While it has the same phenotype as fatal familial insomnia (FFI), it lacks the mutation at codon 178 of the PRNP gene. The initial course of the disease is marked by a significant disturbance of the sleep-wake cycle accompanied by autonomic dysfunction and changes in hormone secretion. Other symptoms, such as ataxia, myoclonia, pyramidal signs, asterixis, and trembling, begin appearing progressively. The most characteristic neuropathological change is atrophy with neuronal loss and gliosis in absence of spongiosis of the anterior ventral and medial dorsal thalamic nuclei and the olivary body.67
PET studies with fluorodeoxyglucose (18F) (or 18F-FDG) performed in patients with sporadic fatal insomnia have revealed thalamic hypometabolism. SPECT studies have demonstrated decreased cerebral blood flow (CBF) to the thalamus and cortex in patients with the same condition.64
- ∘
VV1: This is the molecular subtype that most often displays cortical hyperintensity on MRI scans with no changes in the basal ganglia or thalamus.68 Signs are predominantly located in the cingulate gyrus, insular cortex, and temporal lobes.
- ∘
- •
Subtypes mainly affecting the basal ganglia (Fig. 1):
- ∘
MV2: This subtype is characterised by the appearance of hyperintensities in the basal ganglia and thalamus.63 It is the only subtype in which the pulvinar sign and ‘hockey stick sign’ may appear, meaning that MV2 is sometimes mistaken for vCJD. Cortical involvement in MV2 is limited, but it most typically includes the frontal lobe and cingulate gyrus.
- ∘
VV2: This subtype is the most likely to affect the thalamus and basal ganglia.57 Cortical signal abnormalities are often restricted to fewer than three regions, one of which will typically be the cingulate gyrus.
- ∘
Identification of typical neuroimaging findings in FLAIR and DWI sequences according to the molecular subtype of sCJD.
| Type | Basal ganglia | Thalamus | Cortex | Cerebellum | ||||||
| Impairment | Typical location | |||||||||
| F | P | T | O | I | ||||||
| CC | H | |||||||||
| MM1 | + | − | + | + | + | + | − | |||
| MV1 | + | + | + | + | + | |||||
| MM2 | − | + | + | + | + | |||||
| VV1 | − | − | + | + | + | |||||
| MV2 | + | + | + | + | + | |||||
| VV2 | + | + | + | + | ||||||
CC: cingulate cortex; F: frontal; H: hippocampus; I: insular cortex; O: occipital; P: parietal; T: temporal; +: impairment at this level; −: no impairment at this level.
SPECT and PET studies using 18-FDG in patients with sCJD have, respectively, revealed hypoperfusion and hypometabolism at locations where hyperintensities were detected using a DWI sequence (Fig. 1).69 In the largest published study to date,69 the fundamental finding described by Henkel et al. was hypometabolism in at least 1 region of the temporal or parietal cortex, together with hypometabolism in at least 1 of the following locations: occipital cortex, cerebellum, or subcortical structures (basal ganglia or thalamus). This pattern is not seen in other rapidly evolving dementias. Furthermore, in 50% of the patients studied at the time the PET scan was taken, there were no observable changes on the MRI. This indicates that a PET scan may be useful in early stages of the disease.
Hereditary forms (autosomal dominant inheritance)Familial CJD (fCJD)Hereditary forms account for approximately 15% of all prion diseases. Researchers have described numerous mutations of the PRNP gene in association with fCJD (codons 129, 180, 183, 200, 210, 219, 220, and 232); the most common is E220K. Penetrance is nearly 100%. The condition may be difficult to diagnose considering that its clinical presentation varies and the characteristics typical of prion diseases do not always appear. Normal values for 14-3-3 protein and NSE in the CSF may be detected in up to 25% of patients with fCJD.45 MRI neuroimaging results are very similar to those in sporadic CJD viewed with a DWI sequence. Findings include increased signal intensity in the basal ganglia and cerebral cortex. Signal abnormalities in the caudate nucleus or putamen in the FLAIR sequence are 87% sensitive and 91% specific for forms of fCJD. Hyperintensity in the caudate nucleus in the DWI sequence is 73% sensitive and 100% specific for the disease. The cortical regions in which hyperintensities are the most common are the insular cortex, cingulate gyrus, and the medial region of the frontal, parietal, and occipital lobes (Fig. 1).70
In isolated cases of fCJD, PET scans with 18-FDG have revealed marked hypometabolism in the cerebellum, basal ganglia, thalamus, and cerebral cortex (Fig. 1).71 SPECT imaging shows bilateral hypoperfusion in the thalamus, which in some cases may appear even before changes are detected in a DWI sequence.72 Spectroscopy studies have observed a decrease in the NAA/Cr ratio with increased levels of Mi in the thalamus.73
Gerstmann–Sträussler–Scheinker syndrome (GSS)GSS is characterised by slowly progressive cerebellar ataxia accompanied by cognitive impairment, spastic paraparesis, and extrapyramidal signs. Onset occurs in the sixth or seventh decades of life. The mean duration of the disease varies between 5 and 7 years. It has been linked to a number of genetic alterations affecting PRNP; the most frequent is point mutation P102L.74 The typical neuropathological traits of GSS consist of plaques of PrP in multiple locations plus neurofibrillary neuronal degeneration.
At present, there are no studies containing enough patients to permit analysis of neuroimaging results, and findings from the few studies that have been completed are not homogeneous enough to be useful in diagnosis.74
Regarding functional studies, 1H MRS scans have described a decrease in the NAA/Cr ratio in the frontal lobe, cerebellum, and putamen, even in the absence of other abnormal imaging results.75
PET studies using 18-FDG have shown metabolic decrease in the neocortex, basal ganglia, and/or thalamus. Researchers have observed an increase in 2-(1-{6-[(2-[F-18]fluoroethyl)(methyl)amino]-2-naphthyl}ethylidene)malononitrile ([18F]-FDDNP)76 in association with PrPC deposits, which in turn are linked to symptoms. In PET scans with (11)C-labelled 2-(2-[2-dimethylaminothiazol-5-yl]ethenyl)-6-(2[fluoro]ethoxy)benzoxazole([(11)C]BF-227), the tracer has been described as a non-specific marker of cerebral amyloidosis that may be useful for in vivo detection of PrPSc plaques.77 Lastly, SPECT techniques have been used to detect diffuse hypoperfusion that is predominant in the occipital lobes.78
Fatal familial insomniaAlthough the first clinical description of FFI appeared in 1986, the entity was not listed as a genetic prion disease until 1992. FFI is caused by a mutation in codon 178 of the PRNP gene which elicits the amino acid substitution of asparagine for aspartic acid (D178N). It shares clinical characteristics with the sporadic form of fatal insomnia described previously. FFI is not accompanied by increased levels of 14-3-3 protein in the CSF.
CT and MRI scans have shown non-specific changes amounting to dilation of the ventricles and atrophy of the cerebral and cerebellar cortices.79
PET scans with 18-FDG have revealed extensive hypometabolic areas, predominantly in the thalamus, which are closely correlated to areas with PrPSc deposits.67,80 According to these studies, thalamic and cingulate cortex hypometabolism are characteristic findings in this disease, while impairment in other locations points to disease progression.
ConclusionsNeuroimaging tests constitute an accessible and non-invasive diagnostic tool. Including DWI and FLAIR sequences in study protocols for suspected cases of prion disease has been established for vCJD, and these tests should not be disregarded for other forms of prion disease. Furthermore, functional studies are becoming increasingly important. Although few studies have been published to date, their results indicate that these tools may be useful for performing differential diagnosis and for detecting alterations in early stages.
Conflicts of interestThe authors have no conflicts of interest to declare.
This study has not been presented at the SEN's Annual Meeting or at any other event. It has not received financing from any source.
Please cite this article as: Ortega-Cubero S, et al. Neuroimagen estructural y funcional en las enfermedades priónicas humanas. Neurología. 2013;28:299–308.

Neurología (English Edition) sigue las recomendaciones para la preparación, presentación y publicación de trabajos académicos en revistas biomédicas