





Las prionopatías son un conjunto de enfermedades neurodegenerativas producidas por el acúmulo de una isoforma anormal de la proteína priónica celular (PrPc). Se clasifican en adquiridas, hereditarias y esporádicas. Aunque son muchas las características clínicas, evolutivas y anatomopatológicas que las diferencian, todas ellas tienen en común un curso desfavorable y un pronóstico fatal.
DesarrolloSi bien algunos tipos como el kuru, prácticamente han desaparecido, otras formas como la variante de la enfermedad de Creutzfeldt-Jakob (vECJ) o la iatrogénica siguen vigentes y suponen un reto en la medicina actual. El diagnóstico de certeza se realiza postmortem, salvo en el caso de la vECJ en la que la biopsia amigdalar puede detectar el 100% de los casos. Ello ha supuesto que se definan criterios diagnósticos en función de una probabilidad estadística, que precisa la realización de exámenes complementarios tales como el estudio electroencefalográfico y la detección de la proteína 14-3-3 en el líquido cefalorraquídeo (LCR). Solamente la vECJ ha llegado a incluir en los criterios diagnósticos de la OMS el «signo del pulvinar» en resonancia magnética cerebral (RM). En este artículo se revisan los hallazgos de neuroimagen que han sido descritos para cada tipo de prionopatía en pacientes con un diagnóstico de certeza.
ConclusionesLa finalidad es definir la utilidad de estas pruebas complementarias como una herramienta de apoyo para el diagnóstico de este conjunto de enfermedades neurodegenerativas.
Prion diseases are neurodegenerative disorders resulting from the accumulation of a misfolded isoform of the cellular prion protein (PrPc). They can occur as acquired, sporadic, or hereditary forms. Although prion diseases show a wide range of phenotypic variations, pathological features and clinical evolution, they are all characterised by a common unfavourable course and a fatal outcome.
Review summarySome variants, such as kuru, have practically disappeared, while others, for example the variant Creutzfeldt-Jakob (vCJD) or those attributable to iatrogenic causes, are still in force and pose a challenge to current medicine. There are no definitive pre-mortem diagnostic tests, except for vCJD, where a tonsil biopsy detects 100% of the cases. For this reason, diagnostic criteria dependent on statistical probability have had to be created. These require complementary examinations, such as an electroencephalogram (EEG) or the detection of 14-3-3 protein in cerebrospinal fluid (CSF). Only the pulvinar sign in magnetic resonance imaging (MRI) has been included as a vCJD diagnostic criterion. The present review discusses neuroimaging findings for each type of prion disease in patients with a definitive histopathological diagnosis.
ConclusionsThe aim is to define the usefulness of these complementary examinations as a tool for the diagnosis of this family of neurodegenerative diseases.
Las prionopatías forman un conjunto de enfermedades neurodegenerativas, provocadas por la transformación post-transcripcional de la proteína priónica celular (PrPc) en una isoforma anormal llamada PrPsc. Esta proteína anómala tiene propiedades bioquímicas y biofísicas que la hacen infecciosa en ausencia de ácidos nucleicos1.
La PrPc es una glicoproteína de la membrana celular codificada por el gen PRNP del cromosoma 20. Este gen es polimórfico en el codón 129, pudiendo codificar para el aminoácido valina (V) o metionina (M). La homocigosidad para metionina es un factor de riesgo para el desarrollo de las enfermedades priónicas. La PrPc es sintetizada en el citoplasma celular, transportada a través del retículo endoplásmico y el aparato de Golgi, y posteriormente se localiza en la membrana plasmática, fundamentalmente en la matriz extracelular2. La forma madura de esta proteína está compuesta por 209 aminoácidos con un puente disulfuro3.
La PrPc se expresa desde el comienzo de la embriogénesis. En el individuo adulto está presente en las neuronas del cerebro y la médula espinal, y en menor cuantía en las células gliales del sistema nervioso central (SNC) y en distintos tipos de células del tejido musculoesquelético, cardíaco y linforreticular4. Su función es desconocida aunque se ha postulado que puede intervenir en la diferenciación y neurogénesis de células madre neurales5, la renovación a largo plazo de las células madre del sistema hematopoyético6, la activación de linfocitos7, la neuroprotección frente a la apoptosis y el estrés oxidativo8, la señalización transmembrana9, la formación y el mantenimiento de sinapsis10, la captación de iones de cobre11 y la adhesión a la matriz extracelular. El déficit de esta proteína no produce una patología evidenciable12, mientras que su expresión es un requisito indispensable para la aparición de la enfermedad priónica en el huésped13. El desarrollo de las enfermedades priónicas no se debe solamente al acúmulo de PrPsc, sino también a los cambios que experimenta PrPc, habiéndose establecido diferentes hipótesis para explicar cómo su transformación puede provocar la neurodegeneración14.
Las características comunes a todas las enfermedades priónicas son la afectación exclusiva de la enfermedad al SNC, un período de incubación prolongado y un curso progresivo y uniformemente fatal3. Una de las manifestaciones clínicas más comunes a todas a ellas es el desarrollo de una demencia. Todas ellas comparten también características neuropatológicas entre las que se incluyen: pérdida neuronal, gliosis reactiva, placas amiloideas de proteínas priónicas y presencia de grandes vacuolas que generan el característico aspecto de esponja del cerebro. Clásicamente, las enfermedades priónicas se clasifican en formas adquiridas, hereditarias y esporádicas3. A continuación se describen las manifestaciones clínicas y los principales hallazgos de neuroimagen de las distintas entidades.
DesarrolloFormas adquiridasKuruSe trata de una enfermedad priónica de gran interés histórico descubierta en los años cincuenta. Llegó a alcanzar el rango de epidemia entre algunas tribus de Papúa-Guinea y su transmisión se relacionó con la práctica del canibalismo. Era prácticamente exclusiva en mujeres y niños, ya que los varones no solían participar en el ritual mortuorio y, en caso de hacerlo, solían consumir el músculo y no el cerebro u otras vísceras. «Kuru» quiere decir en el lenguaje tribal «tiritar o temblar», una de las características clínicas fundamentales de esta entidad junto a una ataxia cerebelosa progresiva, coreoatetosis y movimientos oculares anormales. El desarrollo clínico de estos síntomas era precedido de una fase prodrómica caracterizada por cefalea y dolores musculares y articulares. Su período de incubación es prolongado y desde el abandono del canibalismo su incidencia ha disminuido15. Los estudios genéticos realizados muestran que las personas expuestas que no desarrollaban la enfermedad eran heterocigotos para el polimorfismo del codón 120 del gen PRNP. Otro estudio más reciente establece que el polimorfismo 127V también supone un factor protector para desarrollar esta enfermedad priónica16. No se dispone de datos de neuroimagen de esta entidad.
Enfermedad de Creutzfeldt-Jakob iatrogénicaSe produce por la transmisión de la infección de persona a persona en el curso de diversos procedimientos médicos o quirúrgicos. El primer caso se describió en 1974 en un paciente que recibió un transplante corneal de un donante que posteriormente falleció por una ECJ17. Desde entonces se han descrito casos de ECJ tras la utilización de material contaminado en procedimientos neuroquirúrgicos, la colocación de electrodos intracerebrales, la administración de hormonas hipofisarias, el transplante de duramadre o de córnea y, más recientemente, tras transfusiones sanguíneas18.
El diagnóstico de la ECJ se basa en la presencia de una clínica típica compatible que incluye demencia, ataxia, signos piramidales y extrapiramidales, mioclonías y mutismo acinético19 junto con un antecedente de exposición. En un 77% de las ocasiones se detecta un aumento de la proteína 14-3-3 en LCR20. Los hallazgos electroencefalográficos son similares a los de la variedad esporádica con presencia de complejos punta-onda periódicos que, en ocasiones, aparecen de forma preferente en el lugar de inoculación21. La histología muestra los datos típicos anteriormente mencionados, aunque se ha descrito una variante de la forma provocada por injerto de duramadre en la que se aprecian placas de PrP de forma difusa. Esta variante cursa de forma insidiosa, predominando el componente atáxico y la demencia. De forma característica no se aprecian alteraciones electroencefalográficas hasta fases muy tardías19.
Tanto la clínica como el período de incubación varían en función de la vía de inoculación. En el caso de inoculaciones intracerebrales o próximas al SNC, el período de incubación es de meses y predomina la demencia como síntoma fundamental. Cuando la inoculación es periférica predomina un cuadro atáxico que remeda al kuru cuyo período de incubación es de hasta 31 años. La mayoría de los pacientes son homocigotos MM para el codón 129, lo que apoya la importancia de este rasgo genético como marcador de susceptibilidad.
Meissner et al19 estudiaron con RM una serie de 10 pacientes que desarrollaron una ECJ tras implante de duramadre. En estos pacientes se observaron hiperintensidades corticales, afectando en orden descendente de frecuencia al giro cingulado y los lóbulos frontal, temporal y parietal en secuencias DWI (diffusion-weighted Imaging) y FLAIR (fluid-attenuated inversion recovery). La alteración de la corteza occipital era menos frecuente y los ganglios basales y el tálamo también se encontraron afectados en un número elevado de pacientes, aunque en esta última estructura sin la aparición del «signo del pulvinar» o del «palo de hockey», característicos de la nueva variante de la ECJ.
En los casos de ECJ iatrogénica tras transplante corneal se han descrito hallazgos similares. En las secuencias DWI y FLAIR se han observado áreas restrictivas extensas en la corteza parieto-occipital y los lóbulos frontotemporales, junto con aumento de señal en el putamen de forma bilateral y en la cabeza del núcleo caudado unilateralmente22.
En el caso de pacientes con ECJ provocada por la administración de hormona de crecimiento se han descrito las mismas alteraciones. No obstante, cabe destacar en estos casos la afectación del cerebelo, que es poco frecuente en otros tipos23 y cuyo hallazgo con una clínica compatible podría apoyar el diagnóstico de esta entidad.
Los estudios con espectroscopia no han llegado a establecer un patrón estándar para esta entidad si bien se ha comprobado que las áreas en las que se aprecia una disminución del pico de NAA corresponden con zonas de muerte neuronal progresiva22,24.
Nueva variante de la enfermedad de Creutzfeldt-JakobLa vECJ o «mal de las vacas locas» fue descrita por primera vez en 1996. El agente causal es el mismo que el de la encefalopatía espongiforme bobina25. La vECJ afecta a pacientes más jóvenes que las formas esporádicas, y la mediana de supervivencia es de 14 meses. Recientemente se ha descrito su presentación familiar26. En el curso clínico inicial predominan los síntomas psiquiátricos, con alteraciones en el comportamiento, cambios en el estado de ánimo, irritabilidad y aislamiento social. Posteriormente, el 50% de los pacientes presenta síntomas sensitivos en forma de disestesias en las extremidades y la cara, siendo muy característico el dolor facial. Conforme la enfermedad evoluciona, aparece ataxia, asociada a movimientos involuntarios que pueden ser coreicos, distónicos o mioclónicos, que recuerdan al kuru. Finalmente existe un deterioro cognitivo progresivo que da lugar posteriormente a un mutismo acinético27. El EEG muestra cambios inespecíficos y la proteína 14-3-3 se detecta en el 50-60% de los casos, con una especificidad superior al 94%. En cuanto a la anatomía patológica, destaca la presencia de placas de PrPsc rodeadas de cambios espongiformes llamadas «placas floridas»28. Al igual que otras enfermedades priónicas, en la vECJ el diagnóstico definitivo se establece tras el examen neuropatológico. Sin embargo, en este caso, dado que puede realizarse un estudio histológico de las amígdalas (la PrPsc se detecta en el 100% de los casos) puede llegarse a un diagnóstico de certeza con el sujeto vivo.
La vECJ es un ejemplo claro de cómo las pruebas de imagen han ido cobrando una mayor importancia, hasta el punto de que el «signo del pulvinar» está incluido en los criterios diagnósticos redactados por la OMS para esta enfermedad (tabla 1). El «signo del pulvinar» consiste en un aumento de señal simétrico en ambos núcleos pulvinares del tálamo con respecto a la corteza cerebral y la parte anterior del putamen que se observa en RM. Puede detectarse en secuencias T2, densidad protónica (DP), FLAIR y DWI. Tiene una sensibilidad para el diagnóstico de la vECJ entre el 78 y el 90% y una especificidad cercana al 100%29. Otro signo característico es el llamado «palo de hockey» en el que el «signo del pulvinar» se combina con aumento de señal en el núcleo dorsomedial del tálamo29 y aparece en el 56% de los casos (fig. 1). Estos hallazgos encontrados en RM parecen ser el resultado de una astrocitosis en el núcleo pulvinar29,30, aunque en esta localización y en el núcleo dorsomedial se produce además de gliosis, pérdida neuronal y rarefacción del neuropilo. Los cambios espongiformes, sin embargo, son más prominentes en el núcleo caudado y en el putamen. Estos cambios no se han correlacionado con un patrón específico en la RM. Recientemente se ha descrito el «signo de la cruz» en un caso de vECJ. Consiste en una hiperintesidad pontina en forma de cruz, característica de la atrofia multisistémica31. Así mismo, en un pequeño número de casos se ha descrito un aumento de señal en la sustancia gris periacueductal. El diagnóstico diferencial de la alteración de señal de RM observada en el tálamo de estos pacientes ha de establecerse con las prionopatías esporádicas. En estas existe también una afectación talámica sin el «signo del pulvinar» y la hiperintensidad en el tálamo suele ser menor que la detectada en la cabeza del caudado y es de carácter más asimétrico32. En el diagnostico diferencial deben también incluirse entidades como la hipertensión intracraneal, la enfermedad por el arañazo de gato33, el síndrome de Alpers34, síndromes paraneoplásicos35 y encefalitis postinfecciosas36.
Criterios diagnósticos ECJ
| I: |
| A. Alteración neuropsiquiátrica progresiva |
| B. Duración de la enfermedad > 6 meses |
| C. No existe diagnóstico alternativo que explique los síntomas |
| D. No hay antecedente de potencial exposición iatrogénica |
| E. No hay evidencia de que se trate de una forma familiar |
| II: |
| A. Síntomas psiquiátricos tempranos |
| B. Síntomas sensitivos dolorosos persistentes |
| C. Ataxia |
| D. Mioclonía, corea o distonía |
| E. Demencia |
| III: |
| A. EEG no muestra los signos típicos de la forma esporádica (o no se realiza) |
| B. RM muestra aumento de seña bilateral y simétricos del pulvinar |
| IV: |
| A. Biopsia lingual positiva |
| Definitivo: IA y confirmación neuropatológicas para vECJ |
| Probable: I y 4/5 II y IIIA y IIIB; I y IVA |
| Posible: I y 4/5 de II y IIIA |
 PET-FDG muestra hipometabolismo talámico bilateral y hemisférico izquierdo. (B) FLAIR sin alteraciones relevantes. (C) Aumento de señal en tálamo dorsomedial bilateral en secuencias de difusión. Caso de eECJ. (A) PET-FDG muestra hipometabolismo en núcleos caudado, tálamo y región anterior del putamen. Hipometabolismo región del polo anterior y frontal dorso-medial de predominio izquierdo. (B) Hiperintensidad en la rodilla de la cápsula interna izquierda y ganglios de la base bilateralmente en secuencias FLAIR. (C) Hallazgos similares en secuencias de difusión con hiperintensidad también en tálamo y córtex cingular. Probable caso de eECJ (subtipo MM2). (A) PET-FDG muestra disminución generalizada de la actividad cortical y de ambas regiones talámicas. (B) Hiperintensidad cortical bihemisférico de predominio izquierdo en secuencias FLAIR. (C) Hallazgos similares en secuencias de difusión. Caso de fECJ. (A) PET-FDG evidencia hipometabolismo de la región talámica y ganglios de la base de predominio hemisférico derecho con compromiso de caudado bilateralmente.")
Caso de vECJ. (A) PET-FDG muestra hipometabolismo talámico bilateral y hemisférico izquierdo. (B) FLAIR sin alteraciones relevantes. (C) Aumento de señal en tálamo dorsomedial bilateral en secuencias de difusión. Caso de eECJ. (A) PET-FDG muestra hipometabolismo en núcleos caudado, tálamo y región anterior del putamen. Hipometabolismo región del polo anterior y frontal dorso-medial de predominio izquierdo. (B) Hiperintensidad en la rodilla de la cápsula interna izquierda y ganglios de la base bilateralmente en secuencias FLAIR. (C) Hallazgos similares en secuencias de difusión con hiperintensidad también en tálamo y córtex cingular. Probable caso de eECJ (subtipo MM2). (A) PET-FDG muestra disminución generalizada de la actividad cortical y de ambas regiones talámicas. (B) Hiperintensidad cortical bihemisférico de predominio izquierdo en secuencias FLAIR. (C) Hallazgos similares en secuencias de difusión. Caso de fECJ. (A) PET-FDG evidencia hipometabolismo de la región talámica y ganglios de la base de predominio hemisférico derecho con compromiso de caudado bilateralmente.
Existen muy pocos estudios de neuroimagen funcional en pacientes con vECJ26 (fig. 1). Mediante tomografía computerizada de emisión de fotón único (SPECT) con Tc-99m hexametil propileno amino oxima (Tc-99m HMPAO) se han objetivado alteraciones inespecíficas, tales como hipoperfusión cortical difusa o, por el contrario, más localizada como en región temporoparietal izquierda37. Los estudios de RM con espectroscopia han descrito un aumento de mioinositol (Mi) y una disminución de NAA en el tálamo, y en menor medida en núcleo caudado de estos pacientes38.
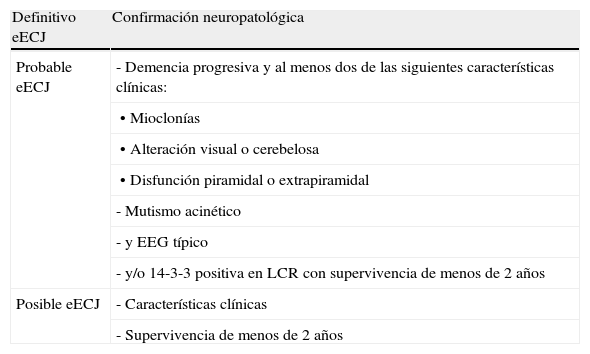
Formas esporádicasEnfermedad de Creutzfeldt-Jakob esporádica (eECJ)Esta enfermedad representa el 85% de los casos de prionopatías humanas. El pico de mayor incidencia se produce en la séptima década de la vida. Los síntomas prodrómicos son inespecíficos e incluyen pérdida de peso, alteraciones del sueño, trastornos de conducta o cambios de personalidad, así como síntomas visuales mal precisados. La marcha y el lenguaje se afectan también en las fases tempranas. Posteriormente aparece una demencia rápidamente progresiva que puede acompañarse de ataxia cerebelosa y mioclonías. La enfermedad finaliza en mutismo acinético con mioclonías y el fallecimiento de los pacientes tras una mediana de supervivencia de 6 meses. No existe una prueba diagnóstica específica, por lo que se siguen unos criterios de probabilidad (tabla 2) y la certeza diagnóstica sólo se establece tras la confirmación neuropatológica.
Criterios diagnósticos ECJ
| Definitivo eECJ | Confirmación neuropatológica |
| Probable eECJ | - Demencia progresiva y al menos dos de las siguientes características clínicas: |
| • Mioclonías | |
| • Alteración visual o cerebelosa | |
| • Disfunción piramidal o extrapiramidal | |
| - Mutismo acinético | |
| - y EEG típico | |
| - y/o 14-3-3 positiva en LCR con supervivencia de menos de 2 años | |
| Posible eECJ | - Características clínicas |
| - Supervivencia de menos de 2 años |
Los primeros estudios de RM realizados a pacientes con eECJ mostraron la presencia de una hiperintensidad simétrica en los ganglios basales (caudado y putamen) en las secuencias potenciadas en T2 y DP. Este hallazgo tiene una sensibilidad del 67% y una especificidad del 93% para el diagnóstico de la enfermedad39. Posteriormente se ha comprobado la superioridad de las secuencias FLAIR y sobre todo de la secuencia DWI39–43 (fig. 1). Esta técnica tiene una mayor capacidad de detección de esta enfermedad44 no solamente frente a T2, DP y FLAIR, sino también respecto al EEG (50-78%), y la determinación de las proteínas 14-3-3 (84-94%) y enolasa específica de neuronas (NSE) (73%) en el LCR45,46. La utilización conjunta de FLAIR y DWI aumenta la sensibilidad diagnóstica de la RM al 91% y la especificidad al 95%40. Recientemente Lodi et al43 han sugerido la conveniencia de añadir a los protocolos actuales de RM para diagnóstico de la eECJ el estudio talámico mediante espectroscopía de RM protónica (1H-MRS). Estos autores describen que la combinación del cociente talámico NAA/Cr en la secuencia 1H-MRS (punto de corte ≤ 1,21) con una hiperintensidad en estriado/córtex en secuencias DWI permite clasificar adecuadamente al 93% de los pacientes con eECJ.
En los pacientes con eECJ se definen dos patrones fundamentales en las secuencias DWI. El primero que aparece en dos tercios de los pacientes se caracteriza por la hiperintensidad de regiones corticales y estructuras subcorticales, fundamentalmente el estriado y tálamo medial y/o posterior. El segundo se define por la hiperintensidad exclusiva a nivel cortical. A estos patrones se les atribuyen implicaciones pronósticas, con mayor mediana de supervivencia en caso de afectación exclusivamente cortical41. En un 7% de los pacientes con eECJ no se encuentra ningún cambio de señal y, en un 2% la hiperintensidad se limita de forma exclusiva a los ganglios basales.
En el grupo con afectación cortical exclusiva en secuencia DWI, el 89% presentan afectación de al menos tres áreas corticales, incluyendo habitualmente los lóbulos parietal y frontal (78%). En secuencias FLAIR la afectación cortical es menos extensa, aunque muestra una mayor sensibilidad para detectar el aumento de señal en el hipocampo. En ambas secuencias destaca la falta de afectación de las áreas corticales somatosensorial y visual primarias40,41.
Recientemente Zerr et al47 han propuesto la creación de unos nuevos criterios diagnósticos para la eECJ que aportan una sensibilidad del 98%. Éstos, además de considerar el EEG y la detección de proteína 14-3-3 en LCR incluyen la presencia en FLAIR o DWI de una hiperintensidad en el núcleo caudado y putamen o en al menos dos regiones corticales.
Entre las posibles causas de falsos positivos se han descrito casos de encefalitis48–50, hipoxia51, epilepsia52, intoxicación por monóxido de carbono53, hipoglucemia, enfermedad de Wilson54, enfermedades mitocondriales55, leucoencefalopatía posterior reversible56, corea de Huntington, así como otras entidades que cursan con alteración cognitiva con los que habría que hacer diagnóstico diferencial57.
Las alteraciones de RM en DWI se corresponden con los cambios espongiformes ya que el pequeño tamaño de las vacuolas condiciona una disminución de la difusión del agua libre58. El hecho de que en el comienzo de la enfermedad la característica neuropatológica esencial sean las vacuolas, explica por qué la secuencia DWI es la más sensible en el diagnóstico de esta entidad. Con la progresión de la enfermedad los cambios glióticos se hacen más prominentes y pueden ser detectados mejor en las secuencias T2 y FLAIR.
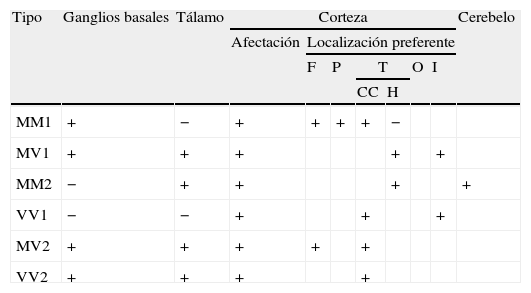
La combinación del genotipo en el codón 129 junto con el tipo molecular de la proteína en función del comportamiento de la misma tras la digestión parcial con proteinasa K (tipos 1 y 2), da lugar a 6 subtipos moleculares de esta enfermedad con diferente neuropatología, clínica y alteraciones en la neuroimagen. Los subtipos MM1 y MV1 son los más frecuentes y suponen el 75% de las prionopatías esporádicas59. En general son formas de rápida evolución que se presentan típicamente con deterioro cognitivo y con alteraciones visuales corticales (variante Heidenhain60). Existen complejos pseudoperiódicos en el EEG y la determinación de la proteína 14-3-3 en LCR es positiva en más del 96% de las ocasiones45,61. Las variantes MM2 y VV1 o corticales se caracterizan por el inicio a una edad temprana. La duración de la enfermedad suele ser mayor y la progresión es relativamente lenta hacia una demencia con desorientación espacial, afasia o apraxia tempranas62. En esas formas suele existir un aumento de los niveles de proteína 14-3-3 en el LCR, mientras que el EEG no muestra las alteraciones típicas. El subtipo MV2 cursa con demencia, ataxia y síndrome rígido acinético. Las mioclonías pueden surgir de forma excepcional en estadios finales. En estos pacientes no se detectan valores alterados de proteína 14-3-3 y el EEG suele ser normal o con alteraciones inespecíficas, por lo que las pruebas de neuroimagen pueden ser de especial utilidad63. En el subgrupo de pacientes VV2 destaca como síntoma inicial la ataxia cerebelosa, aunque la demencia también se describe como signo precoz en un porcentaje significativo de los pacientes. Ocasionalmente estos pacientes muestran las alteraciones típicas en el EEG siendo más útil para el diagnóstico la determinación de la proteína 14-3-3 en el LCR45.
A continuación describiremos los hallazgos de neuroimagen más característicos en secuencias FLAIR y DWI de la eECJ de acuerdo con los diferentes subtipos moleculares44 (tabla 3).
- •
Afectación cortical y de los ganglios basales:
- ∘
MM157: es frecuente la hiperintensidad en los ganglios basales. En la mitad de los pacientes existe una hiperintensidad extensa cortical que afecta fundamentalmente a los lóbulos frontal, parietal y giro cingular. Es característica la falta de hiperintensidad del hipocampo y tálamo.
- ∘
MV157: existe una hiperintensidad frecuente de los ganglios basales y de la corteza, con la inclusión característica de la ínsula y el hipocampo. A diferencia del MM1, con el que comparte fenotipo, es más frecuente la afectación en corteza y tálamo.
- ∘
- •
Afectación predominantemente cortical:
- ∘
MM2 (fig. 1): se ha descrito una afectación cortical extensa que incluye los lóbulos temporales, siendo característico el aumento de señal del hipocampo y el cerebelo, lo que la diferencia del subtipo MM1. Las alteraciones en los ganglios basales son limitadas, de forma que la ausencia de hiperintensidad a este nivel apoya el diagnóstico. Dentro de este subtipo se han definido las variantes cortical, corticotalámica y talámica64. A esta última también se le denomina insomnio esporádico fatal65.
El insomnio esporádico fatal fue descrito en el año 1999 inicialmente como otra entidad clínica66. Es fenotípicamente igual al insomnio familiar fatal, pero sin la mutación en el codón 178 del gen de la proteína priónica PRNP. Cursa inicialmente con una importante alteración del ritmo sueño-vigilia acompañada de disautonomía y alteración de la secreción hormonal. Progresivamente van apareciendo otros síntomas como ataxia, mioclonias, signos piramidales, asterixis y temblor. La alteración neuropatológica más característica es la atrofia con pérdida neuronal y gliosis, pero sin espongiosis de los núcleos ventral anterior y dorsal medio del tálamo, y de la oliva bulbar67.
En estudios con PET de 18-F-fluorodeoxiglucosa (18F-FDG) realizados en pacientes con insomnio esporádico fatal se ha observado un hipometabolismo en tálamo. En ese mismo tipo de pacientes los estudios con SPECT han mostrado una disminución del flujo sanguíneo cerebral (FSC) en tálamo y corteza64.
- ∘
VV168: es el subtipo molecular que con mayor frecuencia muestra hiperintensidad cortical en RM sin afectación de los ganglios basales ni el tálamo. Predomina la localización en giro cingulado, ínsula y lóbulos temporales.
- ∘
- •
Afectación predominante en los ganglios basales (fig. 1):
- ∘
MV263: se caracteriza por la aparición de hiperintensidad en ganglios basales y tálamo. Es en el único subtipo en el que pueden aparecer los «signo del pulvinar» y del «palo de hockey», por lo que se puede confundir con vECJ. En MV2 la afectación cortical es limitada y con mayor frecuencia incluye al lóbulo frontal y al giro cingular.
- ∘
VV257: es la forma que con mayor frecuencia afecta a tálamo y ganglios basales. La alteración de señal cortical con frecuencia está restringida a menos de tres regiones incluyendo de forma característica el giro cingulado.
- ∘
Localización de hallazgos de neuroimagen característicos en secuencias FLAIR y DWI en eECJ en función del subtipo moleculal
| Tipo | Ganglios basales | Tálamo | Corteza | Cerebelo | ||||||
| Afectación | Localización preferente | |||||||||
| F | P | T | O | I | ||||||
| CC | H | |||||||||
| MM1 | + | − | + | + | + | + | − | |||
| MV1 | + | + | + | + | + | |||||
| MM2 | − | + | + | + | + | |||||
| VV1 | − | − | + | + | + | |||||
| MV2 | + | + | + | + | + | |||||
| VV2 | + | + | + | + | ||||||
CC: circunvolución cingular; F: frontal; H: hipocampo; I: ínsula; O: occipital; P: parietal; T: temporal; +: afectación a este nivel; −: ausencia de afectación a este nivel.
En estudios de SPECT y PET con 18-FDG en pacientes con eECJ se ha observado hipoperfusión e hipometabolismo, respectivamente, en la misma localización en la que existe una hiperintensidad en secuencias DWI en RM69 (fig. 1). Henkel et al, en el estudio con mayor número de pacientes publicado hasta el momento69, describen como hallazgo fundamental un hipometabolismo en al menos una región cortical temporal o parietal, junto con hipometabolismo en al menos otra localización de las siguientes: corteza occipital, cerebelo o estructura subcorticales (ganglios basales o tálamo). Este patrón no se aprecia en otras demencias de rápida evolución. Por otra parte en el 50% de los pacientes estudiados en el momento de la realización de la PET no se apreciaron alteraciones en RM, por lo que podría ser una exploración útil en fases tempranas de la enfermedad.
Formas hereditarias (autosómicas dominantes)ECJ familiar (fECJ)Estas formas suponen aproximadamente el 15% de todas las enfermedades priónicas. Se han descrito diversas mutaciones del gen PRNP asociadas a esta patología (codones 129, 180, 183, 200, 210, 219, 220, 232), siendo la más frecuente la E220K. La penetrancia es cercana al 100%. Su diagnóstico puede ser difícil ya que la presentación clínica varía y las características típicas de las enfermedades priónicas no están siempre presentes. Se ha estimado que hasta en un 25% de los pacientes pueden detectarse valores normales en las proteínas 14-3-3 y NSE en LCR45. Los hallazgos de neuroimagen son muy similares a los detectados en las formas esporádicas en la secuencia DWI de RM, con la que se ha visto aumento de señal en los ganglios basales y en la corteza cerebral. La alteración de señal en el núcleo caudado o en el putamen en la secuencia FLAIR descrita tiene una sensibilidad del 87% y una especificidad del 91% en las formas de fECJ. En la secuencia DWI la hiperintensidad del núcleo caudado tiene una sensibilidad del 73% y una especificidad del 100%. Las regiones corticales en las que se observa aumento de señal con más frecuencia son la ínsula, el giro cingulado, la región medial de los lóbulos frontal, parietal y occipital70 (fig. 1).
En casos aislados con esta patología la PET de 18-FDG ha mostrado un hipometabolismo marcado en el cerebelo, ganglios basales, tálamo y corteza cerebral71 (fig. 1). En la SPECT existe una hipoperfusión bilateral en el tálamo, que incluso en algunos casos llega a preceder a las alteraciones detectadas con la secuencia DWI72. Por espectroscopía se ha objetivado una disminución del cociente NAA/Cr con aumento de los niveles de MI en el tálamo73.
Síndrome de Gerstmann-Sträussler-Scheinker (SGSS)El SGSS se caracteriza por ataxia cerebelosa lentamente progresiva, de comienzo en la sexta o séptima décadas de la vida, acompañada de deterioro cognitivo, paraparesia espástica y signos extrapiramidales. La duración media de la enfermedad varía entre 5 y 7 años. Se relaciona con diversas alteraciones genéticas en el gen PRNP, siendo la más frecuente la mutación puntual P102L74. Posee características neuropatológicas típicas, consistentes en placas multicéntricas PrP junto con degeneración neuronal de tipo neurofibrilar.
En cuanto a los resultados de neuroimagen, por el momento, no existe ningún estudio con el suficiente número de pacientes, y en los que hay no se obtienen hallazgos homogéneos que puedan ayudarnos en el diagnóstico74.
En cuanto a los estudios funcionales, con 1H-MRS se ha descrito una disminución del cociente NAA/Cr en el lóbulo frontal, el cerebelo y el putamen, a pesar de no existir alteraciones en otras pruebas de imagen75.
En estudios de PET con 18-FDG se ha observado una disminución del metabolismo en la neocorteza, ganglios basales y/o tálamo. En estas regiones se ha objetivado un aumento de la unión de 2-(1-{6-[F-18]fluoroetil)(metil)amino]-2naftil}etilideno)malonitrilo (18-F FDDNP)76, en relación con el depósito de la proteína priónica y a su vez con los síntomas de los pacientes. También se ha descrito el PET con (11)C 2-(2-[2-dimetilaminotiazol-5-yl]etenil)-6-(2-[fluro]etosi)benzoxazol ([(11)C]BF-227) como un marcador inespecífico de amiloidosis cerebral y potencialmente útil para la detección de las placas de PrPsc in vivo77. Por ultimo, la técnicas de SPECT han mostrado una hipoperfusión difusa de predominio en los lóbulos occipitales78.
Insomnio familiar fatal (IFF)Su descripción clínica se realizó en el año 1986, aunque no fue incorporado como una enfermedad priónica genéticamente determinada hasta el año 1992. Se debe a la mutación en el codón 178 del gen PRNP que provoca una sustitución del aminoácido asparagina por ácido aspártico (D178N). Comparte las características clínicas con la forma esporádica de insomnio fatal que hemos mencionado anteriormente. No se acompaña de aumento de la proteína 14-3-3 en LCR.
En TC y RM se han detectado alteraciones inespecíficas consistentes en dilatación de los ventrículos y atrofia de la corteza cerebral y cerebelosa79.
En PET de 18-FDG se ha objetivado un hipometabolismo extenso de predominio en tálamo, correlacionándose muy bien con las zonas de depósito de PrPsc67,80. Según estos estudios el hipometabolismo talámico y del córtex cingular sería un hallazgo característico de esta enfermedad, mientras que la afectación a otros niveles supondría la expresión de la progresión del cuadro.
ConclusionesLas pruebas de neuroimagen suponen una herramienta diagnóstica accesible y no invasiva. La inclusión de las secuencias DWI y FLAIR en los protocolos de estudio ante una sospecha de enfermedad priónica está establecida en los casos de vECJ pero no puede obviarse en otros tipos de prionopatías. Asimismo, cada vez cobran mayor importancia los estudios funcionales. Si bien son pocos los trabajos publicados, los resultados son prometedores tanto como ayuda en el diagnóstico diferencial como por la precocidad de las alteraciones detectadas.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.
El trabajo no se ha presentado en la reunión de la SEN ni en ninguna otra. No se ha realizado con ningún tipo de financiación.

