



Delayed vasospasm has traditionally been considered the most important determinant of poor outcome after subarachnoid haemorrhage (SAH). Consequently, most of the research and therapies are directed towards reducing the incidence of vasospasm (VSP). To date, however, clinical trials based on this strategy have not delivered a definitive treatment for preventing or reducing brain injury after SAH. This fact has caused a paradigm shift in research, which now focuses on early brain injury (EBI) occurring in the first 72hours after SAH. It has also changed the idea of VSP's role in brain damage, and suggests the need for re-evaluating the pathophysiological process of SAH.
DevelopmentThis review examines the current state of knowledge on the pathophysiological mechanisms associated with EBI and summarises the diagnostic options currently available.
ConclusionIt seems that the research approach needs to be changed, so that investigators will focus on prevention of EBI, reduction of secondary brain complications and ultimately, the optimisation neurological outcome.
El vasoespasmo (VSP) ha sido tradicionalmente considerado como el principal determinante de mal pronóstico tras sufrir una hemorragia subaracnoidea (HSA). Como consecuencia, la mayoría de las líneas de investigación y los tratamientos están dirigidos hacia la reducción de la incidencia de dicho VSP. Hasta la fecha, sin embargo, los resultados de los ensayos clínicos basados en esta estrategia no se han traducido en un tratamiento definitivo capaz de prevenir o mejorar la lesión cerebral tras una HSA.
Este hecho ha provocado un cambio de paradigma en el interés investigativo, focalizándolo hacia la lesión cerebral precoz (LCP), que se produce en las primeras 72 horas tras la HSA. Así mismo, ha modificado la visión que se tenía de la responsabilidad del VSP sobre el daño cerebral y sugiere la necesidad de una re-evaluación del proceso fisiopatológico de la HSA.
DesarrolloEsta revisión examina el estado actual del conocimiento de los mecanismos fisiopatológicos relacionados con la LCP y resume las opciones diagnósticas disponibles en la actualidad.
ConclusiónParece necesario cambiar la dirección en la investigación de esta enfermedad, centrándose en la prevención de la LCP, la reducción de las complicaciones cerebrales secundarias y en última instancia, la optimización de los resultados neurológicos.
Subarachnoid haemorrhage (SAH) remains one of the most frequently studied neurological diseases and is responsible for 5% of all cerebral infarcts.1 Some 10% to 12% of all patients die before reaching the hospital, and between 45% and 50% also die within 30 days of the event.2 Of the surviving patients, 30% present incapacitating sequelae and 66% report changes in their quality of life following the haemorrhage.2
In the past, research largely focused on limiting or preventing arterial vasoconstriction in order to combat the high morbidity and mortality associated with SAH. This approach was based on the clinical correlation between appearance of vasospasm (VSP) and neurological impairment. While our knowledge of pathophysiology in VSP has progressed considerably, these advances have not led to the development of clinically effective treatments. Results such as those obtained with clazosentan3 have changed views about the involvement of VSP in brain injury following SAH. With this in mind, it may be necessary to re-evaluate the pathophysiological process of the haemorrhage. It appears that VSP and its clinical consequences should not be regarded as the only causes of poor prognosis following SAH. Furthermore, the literature supports the position that VSP is not a prerequisite for late-onset brain injury or poor clinical prognosis after an SAH.4,5
In recent years, researchers have analysed 2 fundamental phenomena that develop throughout the phase preceding VSP: early brain injury (EBI) and cortical spreading depression (CSD).6,7 Both experimental and clinical experiences have highlighted the significance of this phase and recognised the important role played by transient ischaemia after SAH onset, the changes in the blood-brain barrier (BBB), and the presence of generalised cortical ischaemia after the haemorrhage. Evidence shows that physiological and cellular events in EBI (occurring within the first 72hours after aneurysm rupture) have a substantial influence on patient outcomes. As prognostic factors, they may be considered even more important than VSP itself.8 EBI should therefore be regarded as the main target for SAH research.
This review analyses the current state of knowledge about pathophysiological mechanisms associated with EBI and offers a summary of currently available diagnostic options.
DevelopmentEarly brain injury and its pathophysiological mechanismsOne of the most important advances made in recent years was learning to recognise EBI after SAH, along with the consequences resulting from the initial haemorrhage, and the harmful effects of both processes on the patient's outcome. The term EBI was coined recently to refer to brain lesions occurring in the first 72hours after SAH, before VSP develops.
Recent studies have focused on the use of therapeutic agents able to lessen EBI. Examples include statins,9 which can attenuate the caspase-dependent apoptosis pathway; melatonin,10 a neurohormone with an antioxidant mechanism that mitigates cerebral oedema and increases survival in experimental models; and hyperbaric oxygen,11 which lessens EBI following experimental SAH by counteracting the harmful effects of CSD and regulating genes and protein systems involved in the response to oxidative stress. This evidence indirectly suggests that multiple pathways and mechanisms participate in EBI pathophysiology and urges us to undertake more in-depth research on EBI.
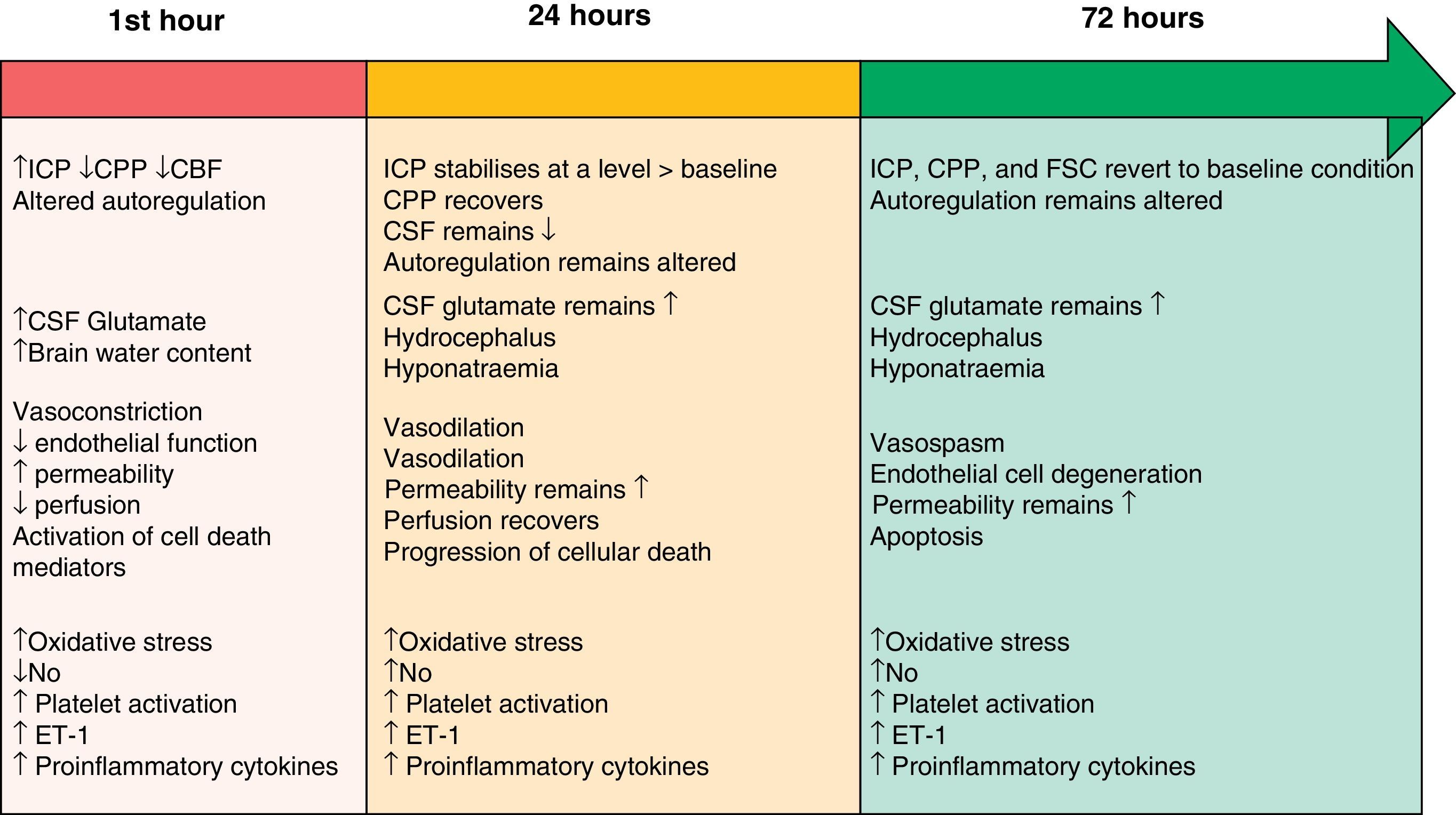
The two pathophysiological mechanisms that we describe in the following sections are clearly interrelated and occur simultaneously. However, the text presents them in separate sections to facilitate comprehension. Their progression timeline during the EBI phase is summarised in Fig. 1.

Timeline showing the different pathophysiological mechanisms intervening during the EBI phase after SAH. Modified from Sehba et al.26 ET-1: endothelin-1; CBF: cerebral blood flow; CSF: cerebrospinal fluid; NO: nitric oxide; ICP: intracranial pressure; CPP: cerebral perfusion pressure.
Initial brain injury is caused by the mechanical effect of blood entering the subarachnoid space.3,12 This traumatic injury causes constriction and compression of the arteries adjacent to the haemorrhagic focus, as well as displacement of cerebrospinal fluid (CSF). These processes elicit an overall rise in intracranial pressure (ICP); the magnitude of the change is related to the volume of blood, the extent to which CSF circulation is obstructed, and the spread of vasodilation in distal cerebral arteries.13 The severity of the increase in ICP is associated with changes in cerebral metabolism, the decrease in cerebral blood flow (attributed to the brief period of no-reflow caused by elevated ICP and the drop in cerebral perfusion pressure [CPP]) and the development of cerebral ischaemia.4,14,15 Consequently, ICP levels are often used to predict SAH prognosis.16 The ICP and CPP fluctuate in the early hours following a haemorrhage, and return to their baseline levels several hours to days later.17 On the other hand, cerebral blood flow (CBF) may either recover or remain low, and the brain's ability to autoregulate will still be affected 72hours after symptom onset.18
Although large cerebral blood vessels contribute to vascular resistance, they have no significant effect on CBF.19 The contraction of large arteries and the dilation of arterioles give rise to a drop in microvascular pressure without eliciting changes in the CBF. This situation shows that microvascular pressure is independently regulated, and it contradicts the idea that large blood vessels contribute significantly to the decrease in CBF. Researchers have identified changes in the anatomical structure of cerebral microvessels that are important enough to provoke functional impairment following SAH.20 It is believed that the brain's vascular autoregulation mechanism fails to compensate for decreased vascular diameter on the microvascular level, and that this failure increases the risk of ischaemia. These changes may explain the origin of cerebral ischaemia in human cases in which angiographic evidence does not show vasoconstriction of large blood vessels.21
InflammationInflammation is a cause–effect reaction that occurs in live tissues in response to all types of lesions. The escaping blood in SAH is responsible for activating a cascade of reactions that cause various vasoactive and proinflammatory factors to be produced in the subarachnoid space. Activation of the inflammatory cascade after SAH involves VCAM-1, ICAM1, and E-selectin, which are the adhesion molecules needed in order for leucocytes to collect in the inflammatory tissues; cytokines (IL-6, IL-1, TNF-α); and complement factors (C3a and C5a) that are able to accelerate erythrocyte lysis and consequently release spasmogenic factors in those cells.22 Clinical studies involving use of anti-inflammatory agents23 have shown that multiple factors are involved in post-SAH inflammation. Studies involving the use of specific IL receptor antagonists and antibodies to block leucocyte/endothelial cell interactions are currently underway.
MicrothrombosisPlatelet activation occurs just a few minutes after SAH and promotes an additional inflammatory mechanism that may exacerbate the secondary brain lesion. Most types of reduced vascular perfusion that have been observed in experimental studies24,25 show platelet aggregates and focal losses of collagen IV. These data indicate that the function of parenchymal microvessels is compromised within 10minutes of SAH onset. They also identify focal microvascular constriction and local accumulation of platelet aggregates in lumina as factors potentially favouring that functional loss.
Oxidative stressOxidative stress is defined as the stress occurring when the physiological balance between pro-oxidants and antioxidants tips in favour of pro-oxidants, eliciting molecular and therefore cellular damage due to oxidation. Recent studies8,26 have provided sufficient evidence to show that oxidative stress plays an important role in EBI. The most important producers of free radicals (ROS or reactive oxygen species) after SAH are release of the superoxide anion, which is stimulated by the presence of oxyhaemoglobin in the CSF; and hydrogen peroxide, produced by the oxidation of haemoglobin to methaemoglobin.27
Reactive oxygen species cause damage to the endothelium and vascular smooth muscle, alter the BBB, and induce apoptosis after SAH.4,28 On the other hand, decreased effectiveness of endogenous antioxidant systems during SAH seems to contribute to progressive neurological impairment in these patients. This statement is supported by studies in which antioxidant therapy provided neuroprotection following an SAH by preserving the BBB,29 preventing cell death,30 and improving patients’ final neurological scores.26
Nitric oxideNitric oxide (NO) plays a fundamental role in cerebral haemodynamics, and any change in NO levels will result in different pathological consequences. NO is produced by the 3 isoforms of nitric oxide synthase (NOS; neuronal, endothelial, and inducible) and its half-life is measured in seconds. Levels of NO decrease, recover, and rise in the first 72hours following SAH.31 Such a variable array of levels leads to changes that range from vasoconstriction, decreased CBF, and platelet aggregation with leucocyte adhesion in the hours following the event, to the appearance of late-onset ischaemic neuronal damage and poor clinical prognosis.32
EndothelinEndothelin-1 (ET-1), a growth factor derived from proinflammatory platelets and cytokines, is a potent vasoconstrictor released by leucocytes and astrocytes in response to the inflammation and early-onset ischaemia present after SAH.33 Many studies have shown high levels of ET-1 in plasma, CSF, and microdialysate after SAH.34 ET-1 is released minutes after haemorrhaging begins, and its levels increase rapidly as NO levels decrease in a process that leads to degenerative morphological changes in vascular walls. This situation promotes cerebral vasoconstriction and provides an additional mechanism related to the pathogenesis of late-onset VSP and late-onset ischaemic neuronal damage.35
Cortical spreading depressionSAH provokes a change in ionic homeostasis which favours vasoconstriction and the appearance of alterations in electrical activity, including cortical spreading depression (CSD). CSD describes a wave of depolarisation in the cortex associated with a net influx of cations (sodium and calcium) and water. If ionic homeostasis is not re-established, cellular oedema may be prolonged (cytotoxic oedema), which can induce cell death. CSD elicits changes in vascular tone, resulting in areas of transient hyperperfusion in healthy tissue (haemodynamic physiological response) through the mechanism of vascular dilation. Alternatively, it causes areas of hypoperfusion in tissues at risk for a progressive lesion (inverse haemodynamic response) by provoking severe microvascular spasm.36 In the second case, the widespread perfusion deficit prolongs the episode of neuron depolarisation. Experimental studies have shown that oxyhaemoglobin, increased extracellular potassium, and decreased levels of NO, glutamate, and ET-1 are all involved in the development of CSD after SAH.37,38 Recent clinical studies39,40 provide direct and unmistakeable electrophysiological evidence as to the presence of CSD in SAH and how it is related to the development of late-onset ischaemic lesions.
Hyponatraemia and hypomagnesaemiaPublished incidence rates of hyponatraemia after SAH range from 10% to 30%.1 The condition frequently appears in patients with aneurysms of the anterior communicating artery, hydrocephalus, and poor initial clinical condition. Hyponatraemia is regarded as an independent risk factor for unfavourable prognosis.
Furthermore, approximately 38% of patients with SAH have low magnesium levels at 48hours after onset of the event. Low magnesium levels are related to vascular dilation and the inhibition of platelet aggregation and ET-1 synthesis, meaning that low magnesium exacerbates EBI. These deficits also contribute to increased intracellular levels of calcium, which is regarded as the main mediator of neuronal death. This knowledge is delivering such new treatment approaches as the MASH-II study,41 which is currently underway.
GlutamateResearchers have observed that both glutamate and glutamate transporters (GT) play an important role in the pathogenesis of ischaemic neurological lesions. Their levels seem to be associated with the intensity of the initial injury. Changes in glutamate, GTs, and neuronal damage after SAH have not yet been fully studied. However, induced SAH in a rat model seems to produce an excessive and prolonged increase in extracellular GT levels and downregulation of GT. These processes are accompanied by increased basilar artery wall thickness and neuronal degeneration in the hippocampus.42
ApoptosisThe initial phenomenon occurring immediately after rupture of the aneurysm is acute CBF arrest, resulting in a state of transient global brain ischaemia which can itself be lethal. In cases in which patients survive such events, secondary ischaemic insult may appear due to changes in the BBB, and it may progress to the point of causing generalised cerebral oedema and/or apoptosis. Cerebral oedema contributes to yet another increase in ICP, resulting in further reduction of the CBF.43 The mechanism of BBB impairment is unclear, but apoptosis has been posited as one possible cause.44 Changes in the BBB and the subsequent formation of brain oedema have been listed as the main predictive factors for cognitive dysfunction following SAH.45 Some authors report that apoptosis is even involved in the formation and rupture of aneurysms, in both animal models and in humans.46,47 While numerous apoptosis activation pathways have been studied, 3 main pathways are recognised.6 These are the death receptor pathway (Fas, TNFR1, and DR 3–5); the caspase-dependent pathway (represented by caspases 3 and 8) and caspase-independent pathway (represented by the apoptosis-inducing factor); and the mitochondrial pathway (represented by cytochrome c and nuclear transcription factor p-53). The targets of these apoptotic pathways are essentially neurons, glial cells, and cerebral vascular cells (smooth muscle and endothelium).
Diagnostic optionsIt is obvious that many of the current diagnostic techniques for SAH are only able to identify physiological changes once neuronal damage is so well-established that it is probably irreversible. Therefore, a sizeable percentage of current research on this topic focuses on identifying markers that enable early detection of the lesions produced by SAH. The two main lines of research in pursuit of this objective explore neuroimaging techniques and biomarkers in bodily fluids.
Neuroimaging techniquesNumerous methods of measuring cerebral perfusion have been used in recent years.48–50 These include positron emission tomography, single-photon emission computed tomography, xenon-enhanced computed tomography, and transcranial Doppler (TCD) imaging. Of the methods listed above, TCD is the most widely used due to being a non-invasive process that yields good results. However, we must be aware of its limitations: the method is operator-dependent, unable to measure CBF, and offers non-specific information regarding the treatment to be used. On the other hand, images obtained by computed tomography are an emerging topic in this field. One such option in CT imaging is CT perfusion, or CTP.51
CTP is being investigated extensively as a method for obtaining early diagnosis of both EBI52 and cerebral VSP,53 with promising results. CT perfusion imaging is able to provide several quantitative parameters for brain haemodynamics, including mean transit time (MTT), cerebral blood volume, and CBF. In practice, visual inspection of CTP maps may be used as a reliable method for assessing cerebral hypoperfusion. These maps may show decreased perfusion in areas with minimal or no VSP according to the angiography. This fact lets us identify ischaemic risk which would otherwise go unnoticed. In fact, findings from different studies employing CTP seem to have a better predictive value than those from studies using CT angiography51 or TCD54 to diagnose late-onset cerebral ischaemic lesions in patients with signs of clinical impairment.
Of all the measurements taken by CTP, mean transit time (MTT) has been identified not only as the most sensitive perfusion parameter for detecting cerebral VSP, but also as an independent predictor of early mortality following SAH.55 In any case, as a parameter within the EBI phase following SAH, MTT seems to have potential for early detection of patients at risk for a poor outcome.
BiomarkersLarge volumes of literature are available on biomarker analysis within the context of different human biological samples and SAH. Distinct techniques and theoretical perspectives have been used to study biomarkers at different historical moments. Each approach evolved from the theoretical understanding of data such as the underlying pathophysiological mechanisms in the development of VSP or in existing EBI.
Techniques including cerebral microdialysis (CMD) and proteomics may be useful for studying the pathogenesis of EBI and VSP following SAH.
Although CMD has delivered contradictory results, it is regarded as a promising method for monitoring patients who have experienced SAH. Research shows that transient decreases in CBF correlate to extracellular increases in glutamate and glycerol, while the lactate/pyruvate ratio is only sensitive after prolonged hypoperfusion.56 Reactive oxygen species, the metabolites derived from NO, and certain astrocytic cytokines and proteins have been harvested by microdialysis and listed as potential VSP biomarkers.57 The future success of CMD will depend largely on the biomarkers selected; biomarkers’ sensitivity, specificity, and predictive values for secondary neurochemical events; and the availability of practical methods for performing chemical analysis of individual markers.
Proteomics delivers a large-scale analysis of all the proteins in a biological sample simultaneously. This advanced detection technique can also be used for objective, non-biased gathering of data enabling identification of biomarkers and/or new treatment targets that may not appear to be linked to any of the disease's pathophysiological processes.
Authors such as Lad et al.58 have reviewed the changes in biomarkers for VSP in CSF. According to their system, biomarkers fall into 3 categories according to their predictive value: (a) markers with auspicious value; (b) candidate markers; (c) noncandidate markers. Category A includes TNF, soluble tumour necrosis factor receptor I, IL-1 receptor antagonist, and neurofilament proteins NFL and NfH. Category B includes apolipoprotein E, F2-isoprostane, NADPH oxidase, and thrombin-antithrombin III complex as markers predicting neurological outcome; E-selectin, lactate, and alpha-II spectrin breakdown products were listed for VSP prognosis. Category C includes glial protein S100B, platelet-derived growth factor, ICAM-1, VCAM-1, and IL-8.
Multimodal neuromonitoringIncreasing numbers of authors state that combining data collected using different techniques can provide doctors with better information, given that each method provides a distinct perspective on cerebral physiology and metabolism. This concept, known as multimodal neuromonitoring, has emerged as an auspicious approach in specific studies of patients with SAH. Examples of the approach could include combined use of conventional monitoring systems (ICP, CTD, etc.) and direct measurements of cerebral microdialysis biomarkers, or using a combination of advanced cerebral and cardiovascular monitoring techniques.59
ConclusionsThe phenomenon of EBI is opening new horizons in SAH research, and it may be a key contributor to high morbidity and mortality rates in SAH. This new research opportunity should foster cooperative efforts among doctors representing basic neurosciences, neurosurgery, neurology, and neurocritical care with a view to achieving better prevention of secondary ischaemic lesions resulting from EBI.
EBI is probably caused by a series of interrelated pathophysiological mechanisms that all yield the same outcome: cell death. We have yet to determine the principal factor or factors in this process. What is clear is that research is delivering spectacular advances; furthermore, since reverting VSP does not seem to improve clinical outcomes, anti-EBI treatments may be a viable treatment option for patients with SAH in the future.
Conflicts of interestThe authors have no conflicts of interest to declare.
We would like to thank Dr Rafael León López and Noa León Muñoz for their invaluable support.
Please cite this article as: Muñoz-Guillén NM, et al. Del vasoespasmo a la lesión cerebral precoz: una nueva frontera en la investigación de la hemorragia subaracnoidea. Neurología. 2013;28:309–16.
