









Late-onset Rasmussen encephalitis (LORE) is a rare, unihemispheric, progressive, inflammatory disorder causing severe neurological dysfunction and drug-resistant epilepsy with onset during late adolescence or adulthood. Due to the scarcity of available evidence, this study aims to improve its clinical characterization and summarize the distinctive features.
DevelopmentThree illustrative cases are presented, including the clinical, neurophysiological, and neuroimaging work-up. Our findings are discussed with reference to previous evidence gathered through a comprehensive search.
The reported patients presented adult onset within a wide age range. The initial clinical manifestation was variable, including refractory focal epilepsy, progressive hemiparesis, and epilepsia partialis continua, in line with previous findings. Progressive hemiatrophy with frontal or posterior predominance in MRI and extensive hypometabolism in functional neuroimaging were documented. Unihemispheric slow background activity and epileptiform discharges progressively developed during the long-term follow-up, as described in the literature. According to the European consensus diagnostic criteria, 2 patients met the Part A and one the Part B criteria. As reported in previous publications, slower neurological decline was observed with immunotherapy.
ConclusionsDespite the wide range of clinical manifestations at onset, overall, LORE presents milder neurological deterioration and responds favorably to immunotherapy, which implies a better prognosis. Further studies are needed to establish the best strategy.
La encefalitis de Rasmussen de inicio tardío (LORE) es un trastorno inflamatorio progresivo unihemisférico infrecuente que causa una grave disfunción neurológica y epilepsia fármacorresistente con debut durante la adolescencia tardía o la edad adulta. Debido a la escasa evidencia disponible, nuestro objetivo es mejorar su caracterización clínica y resumir sus características distintivas.
DesarrolloSe presentan tres casos ilustrativos incluyendo estudios complementarios clínicos, neurofisiológicos y de neuroimagen. Los hallazgos se discuten con la evidencia previa obtenida a través de una búsqueda exhaustiva.
Los pacientes reportados tuvieron un debut en la edad adulta dentro de un amplio rango de edad. La primera manifestación clínica fue variable, incluyendo epilepsia focal farmacorresistente, hemiparesia progresiva o epilepsia parcial continua, en línea con hallazgos previos. La resonancia magnética documentó una hemiatrofia progresiva de predominio frontal o posterior y la neuroimagen funcional un hipometabolismo extenso. Durante el seguimiento a largo plazo, se produjo una lentificación progresiva y desarrollo de actividad epileptiforme de forma unihemisférica, como se describe en la literatura. De acuerdo con los «Criterios Diagnósticos del Consenso Europeo», dos pacientes cumplieron los criterios con la Parte A y uno con la Parte B. En consonancia con publicaciones anteriores, se observó un deterioro neurológico más lento con inmunoterapia.
ConclusionesA pesar de la amplia variedad de manifestaciones clínicas al inicio, en general, LORE tiene un deterioro neurológico más leve y una respuesta favorable a la inmunoterapia, lo que implica un mejor pronóstico. Se necesitan más estudios para aclarar la mejor estrategia terapéutica.
In 1958, Theodore Rasmussen et al.1 reported 3 pediatric patients with intractable seizures and progressive hemiparesis due to chronic unihemispheric encephalitis. Since then, scientists have used the term Rasmussen encephalitis (RE) to refer to this condition.
RE is recognized today as a rare but severe immune-mediated brain disorder that leads to unilateral hemispheric atrophy, progressive neurological dysfunction, and drug-resistant epilepsy.1–3 In Germany, the annual incidence rate of diagnosed RE is estimated at 2.4cases/107 population aged≤18 years, and RE is categorized within the group of rare diseases.4 Although this entity has classically been described as a childhood encephalopathy, about 10% of cases present late onset, in adulthood.5,6 Late-onset RE (LORE) has caused some debate among scientists. In recent years, important new insights have been made in terms of the pathophysiology, diagnosis, and management of the condition, but the available evidence remains limited.7
Although the pathophysiology of this disease remains unknown, several studies point to an immune-mediated mechanism. Regarding cellular immunity, a viral etiology was initially suggested, based on the constituents of the immune reaction (CD8+ T lymphocyte infiltration, microglial nodules, astrocytic activation, vacuolar changes, and capillary proliferation).1 Later, specific attack by cytotoxic T lymphocytes (CD8+) was proposed as the possible mechanism responsible for astrocytic degeneration in RE.8,9 However, more recent studies have proven that CD4 and gamma-delta (γδ) T lymphocytes are also involved.10 There is also evidence of microglial and inflammasome activation,11 as well as participation of humoral immunity, though the role of B-cells and secreted autoantibodies remains controversial.12–14 Numerous autoantibodies have been found in the serum of patients with RE, such as antibodies targeting GluR-3,15 Munc-18,16 alpha-7 nicotinic acetylcholine receptor,17 and AMPA receptor,18 although these are considered only to reflect an immune background rather than playing a primary pathogenic role.19 In addition, variable CSF levels of IgG, CD4+ and CD8+ T cells, granzyme-B, IFNγ, IL-12, and TNF-α have been reported, depending on the stage of the disease, with higher levels during the acute stage.20
This article aims to improve the clinical characterization of LORE and to summarize the existing knowledge, emphasizing the distinctive features of these patients.
DevelopmentMaterial and methodsWe describe 3 illustrative cases and compare their characteristics with the reviewed literature.
In addition, a comprehensive search was conducted of articles published on the MEDLINE (PubMed) database up to August 2022, using the following terms: “RASMUSSEN ENCEPHALITIS” AND [“ADULT-ONSET” OR “LATE-ONSET”], filtering for studies written in English or Spanish. No other specific filters were applied regarding type of study, year of publication, or geographic localization. The inclusion criteria were studies about patients with RE with at least one case of adult- or late-onset forms, considering patients aged 12 years or older.4,7 Most of the available studies were case reports or case series. We excluded studies about different forms of encephalitis other than RE, about childhood-onset RE, or that were not fully available. The literature search is summarized in Fig. 1.
Illustrative casesClinical case 1
A 42-year-old right-handed man with no clinical history of interest was referred to the epilepsy unit. At 32 years of age, he had presented a first episode compatible with a generalized tonic–clonic seizure. Previously, he had presented stereotyped episodes consisting in the appearance of luminous flashes over the right visual hemifield, with occasional episodes of rigidity and jerks in the right leg, lasting seconds. He also complained of a right-sided progressive motor deficit and right visual field impairment. The results of an autoimmune study, including neuronal surface and onconeuronal antibodies, were negative, with the exception of anti-SOX-1. Brain MRI showed left temporo-parieto-occipital atrophy and hyperintensity. Clinical findings, complementary testing, and management are summarized in Table 1 and Fig. 2. Antiepileptic drugs (AED) and immunotherapy were started. Subsequently, the patient presented episodes of motor aphasia lasting one minute after brushing his teeth, so the AED dose was increased. Despite progression of atrophy on brain MRI, after 12 years of follow-up neurological progression has been mild to moderate, with excellent seizure control.
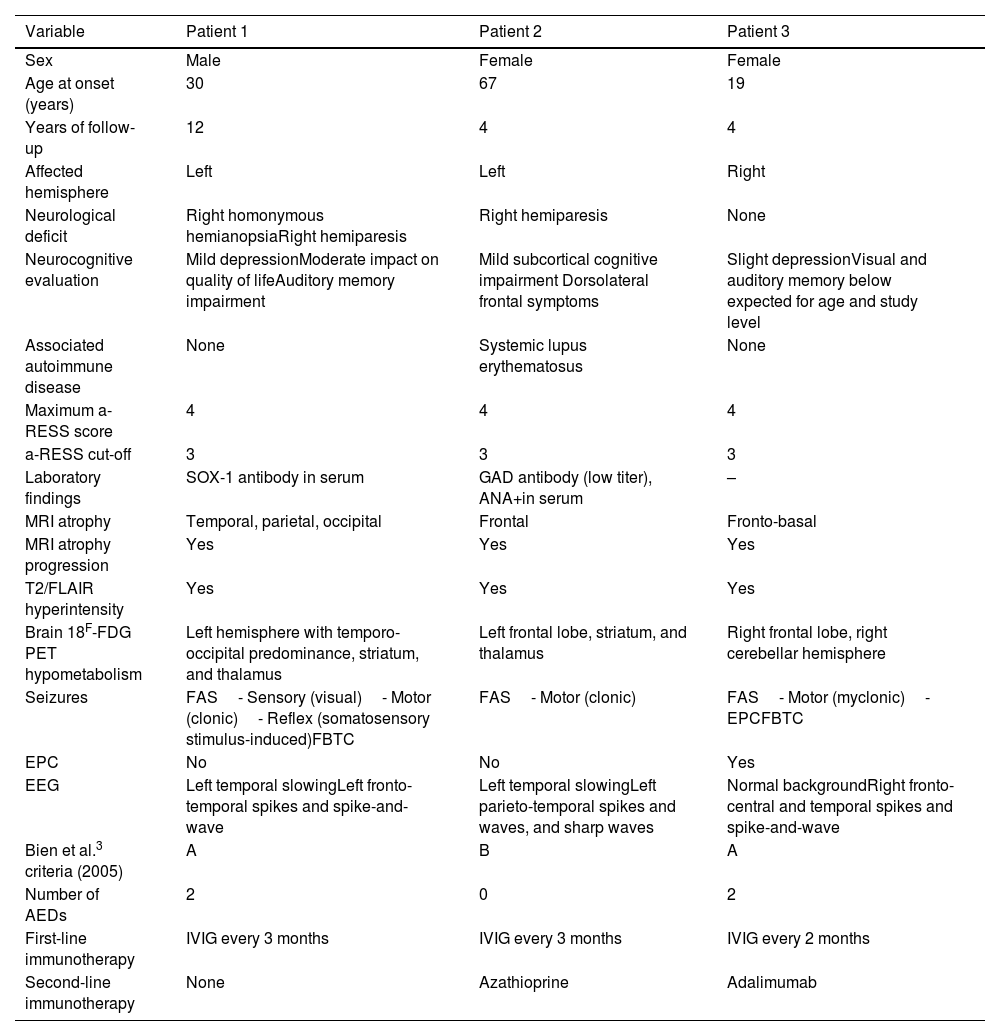
Clinical features and complementary tests.
| Variable | Patient 1 | Patient 2 | Patient 3 |
|---|---|---|---|
| Sex | Male | Female | Female |
| Age at onset (years) | 30 | 67 | 19 |
| Years of follow-up | 12 | 4 | 4 |
| Affected hemisphere | Left | Left | Right |
| Neurological deficit | Right homonymous hemianopsiaRight hemiparesis | Right hemiparesis | None |
| Neurocognitive evaluation | Mild depressionModerate impact on quality of lifeAuditory memory impairment | Mild subcortical cognitive impairment Dorsolateral frontal symptoms | Slight depressionVisual and auditory memory below expected for age and study level |
| Associated autoimmune disease | None | Systemic lupus erythematosus | None |
| Maximum a-RESS score | 4 | 4 | 4 |
| a-RESS cut-off | 3 | 3 | 3 |
| Laboratory findings | SOX-1 antibody in serum | GAD antibody (low titer), ANA+in serum | – |
| MRI atrophy | Temporal, parietal, occipital | Frontal | Fronto-basal |
| MRI atrophy progression | Yes | Yes | Yes |
| T2/FLAIR hyperintensity | Yes | Yes | Yes |
| Brain 18F-FDG PET hypometabolism | Left hemisphere with temporo-occipital predominance, striatum, and thalamus | Left frontal lobe, striatum, and thalamus | Right frontal lobe, right cerebellar hemisphere |
| Seizures | FAS- Sensory (visual)- Motor (clonic)- Reflex (somatosensory stimulus-induced)FBTC | FAS- Motor (clonic) | FAS- Motor (myclonic)- EPCFBTC |
| EPC | No | No | Yes |
| EEG | Left temporal slowingLeft fronto-temporal spikes and spike-and-wave | Left temporal slowingLeft parieto-temporal spikes and waves, and sharp waves | Normal backgroundRight fronto-central and temporal spikes and spike-and-wave |
| Bien et al.3 criteria (2005) | A | B | A |
| Number of AEDs | 2 | 0 | 2 |
| First-line immunotherapy | IVIG every 3 months | IVIG every 3 months | IVIG every 2 months |
| Second-line immunotherapy | None | Azathioprine | Adalimumab |
18F-FDG PET: positron emission tomography with 2-deoxy-2-[fluorine-18] fluoro-d-glucose; AED: antiepileptic drug; a-RESS: Adult-onset Rasmussen Encephalitis Severity Scale; EEG: electroencephalography; EPC: epilepsia partialis continua; FAS: focal aware seizures; FBTC: focal to bilateral tonic–clonic; FLAIR: fluid-attenuated inversion recovery sequence; IVIG: intravenous immunoglobulins; MRI: magnetic resonance imaging.
![Complementary studies of patient 1. (A) EEG (2020). Bipolar circular montage showing discrete diffuse slowing of brain bioelectrical activity. Paroxysmal focality of irritative characteristics of occasional expression (single burst) in the left anterior temporal region with contralateral transmission. (B) EEG (2022). Bipolar circular montage showing slightly slowed background activity with expressive paroxysmal activity in the left fronto-centro-temporal region. Very frequent discharges of spikes and waves at 2–2.5Hz, of 10 seconds’ duration, over the left and central fronto-temporal region (Fp1, F7, Fz), with transmission to contralateral homologous regions. During sleep, the discharges became much more frequent and present greater amplitude and tendency to diffusion. (C) Brain MRI (2010). The FLAIR-weighted image shows prominent cortical atrophy over the left hemisphere with frontal lobe preservation, white matter hyperintensity, and ipsilateral ventricular retraction. (D) Brain MRI (2020). A: Coronal T2-weighted image. B: Axial FLAIR-weighted image. Both images show cortical atrophy associated with extensive gliosis affecting the left parietal and occipito-temporal lobe, associated with ventricular retraction. (E) 18F-FDG PET, co-registered with MRI (2021). Severe hypometabolism or absence of uptake associated with appreciable cerebral atrophy of left temporal and occipital predominance. Hypometabolism and atrophy were also seen in the superior fronto-parietal medial cortex. Moderate hypometabolism was observed in the left temporal pole in the areas with less cortical thickness. The left striatum and left posterior thalamus showed mild hypometabolism. No significant findings were observed in the cerebellum. Results showed the absence of hypermetabolism suggesting inflammatory activity or functional increment by ictal activity. 18F-FDG PET: positron emission tomography with 2-deoxy-2-[fluorine-18] fluoro-d-glucose; EEG: electroencephalography study; FLAIR: fluid-attenuated inversion recovery; MRI: magnetic resonance imaging.](https://static.elsevier.es/multimedia/02134853/unassign/S0213485324001130/v1_202409020413/en/main.assets/gr2.jpeg?xkr=ue/ImdikoIMrsJoerZ+w997EogCnBdOOD93cPFbanNcnxJVYM2BEAEuZ5XZLhi9i85vcbshWVQPfl+0x3NpqwiqkNtigdeFuE9Zyvaz/gsKNfR4ysGK+modMU8s6xaRFTe8NUh3LUk+0JlqzujjzTngRDBXjzKcXNFsxhRyIRupVXqhA2WKaQwENP5ihuSTdMsXYtYPqROenHt8e0adXU42kSRsOrOcUsOtKYTko1sXo1cpyAR87egsyQQA1gPXqX/qWCNcoLYoA1RChn4l18zEw4MdWiMSYGxNAItvBfBk= "Complementary studies of patient 1. (A) EEG (2020). Bipolar circular montage showing discrete diffuse slowing of brain bioelectrical activity. Paroxysmal focality of irritative characteristics of occasional expression (single burst) in the left anterior temporal region with contralateral transmission. (B) EEG (2022). Bipolar circular montage showing slightly slowed background activity with expressive paroxysmal activity in the left fronto-centro-temporal region. Very frequent discharges of spikes and waves at 2–2.5Hz, of 10 seconds’ duration, over the left and central fronto-temporal region (Fp1, F7, Fz), with transmission to contralateral homologous regions. During sleep, the discharges became much more frequent and present greater amplitude and tendency to diffusion. (C) Brain MRI (2010). The FLAIR-weighted image shows prominent cortical atrophy over the left hemisphere with frontal lobe preservation, white matter hyperintensity, and ipsilateral ventricular retraction. (D) Brain MRI (2020). A: Coronal T2-weighted image. B: Axial FLAIR-weighted image. Both images show cortical atrophy associated with extensive gliosis affecting the left parietal and occipito-temporal lobe, associated with ventricular retraction. (E) 18F-FDG PET, co-registered with MRI (2021). Severe hypometabolism or absence of uptake associated with appreciable cerebral atrophy of left temporal and occipital predominance. Hypometabolism and atrophy were also seen in the superior fronto-parietal medial cortex. Moderate hypometabolism was observed in the left temporal pole in the areas with less cortical thickness. The left striatum and left posterior thalamus showed mild hypometabolism. No significant findings were observed in the cerebellum. Results showed the absence of hypermetabolism suggesting inflammatory activity or functional increment by ictal activity. 18F-FDG PET: positron emission tomography with 2-deoxy-2-[fluorine-18] fluoro-d-glucose; EEG: electroencephalography study; FLAIR: fluid-attenuated inversion recovery; MRI: magnetic resonance imaging.")
Complementary studies of patient 1. (A) EEG (2020). Bipolar circular montage showing discrete diffuse slowing of brain bioelectrical activity. Paroxysmal focality of irritative characteristics of occasional expression (single burst) in the left anterior temporal region with contralateral transmission. (B) EEG (2022). Bipolar circular montage showing slightly slowed background activity with expressive paroxysmal activity in the left fronto-centro-temporal region. Very frequent discharges of spikes and waves at 2–2.5Hz, of 10 seconds’ duration, over the left and central fronto-temporal region (Fp1, F7, Fz), with transmission to contralateral homologous regions. During sleep, the discharges became much more frequent and present greater amplitude and tendency to diffusion. (C) Brain MRI (2010). The FLAIR-weighted image shows prominent cortical atrophy over the left hemisphere with frontal lobe preservation, white matter hyperintensity, and ipsilateral ventricular retraction. (D) Brain MRI (2020). A: Coronal T2-weighted image. B: Axial FLAIR-weighted image. Both images show cortical atrophy associated with extensive gliosis affecting the left parietal and occipito-temporal lobe, associated with ventricular retraction. (E) 18F-FDG PET, co-registered with MRI (2021). Severe hypometabolism or absence of uptake associated with appreciable cerebral atrophy of left temporal and occipital predominance. Hypometabolism and atrophy were also seen in the superior fronto-parietal medial cortex. Moderate hypometabolism was observed in the left temporal pole in the areas with less cortical thickness. The left striatum and left posterior thalamus showed mild hypometabolism. No significant findings were observed in the cerebellum. Results showed the absence of hypermetabolism suggesting inflammatory activity or functional increment by ictal activity. 18F-FDG PET: positron emission tomography with 2-deoxy-2-[fluorine-18] fluoro-d-glucose; EEG: electroencephalography study; FLAIR: fluid-attenuated inversion recovery; MRI: magnetic resonance imaging.
A 67-year-old woman with a medical history of hypertension was referred due to progressive motor impairment of the right limbs. The initial contrast MRI study showed a frontal lesion; therefore, a brain biopsy was performed, showing moderate chronic inflammation of perivascular and parenchymal distribution, with an increase in CD8+ T lymphocytes and astrocytosis. No microglial nodules were observed, but the study detected granzyme B immunoreactivity (Fig. 3). Blood and CSF study findings were unremarkable, with the exception of ANA and low-titer GAD serum antibodies. Brain MRI showed left frontal hyperintensity and frontoparietal atrophy. As the patient met the Part B criteria for RE, first- and second-line immunotherapy were initiated. Three years after onset, she presented occasional episodes of involuntary movements in the right hand, consistent with focal motor seizures. Neuroimaging detected progressive cerebral hemiatrophy. Clinical findings and complementary tests are summarized in Table 1 and Fig. 4. Interestingly, a reddish skin lesion was biopsied 4 years later, with findings compatible with systemic lupus erythematosus (SLE). After this period, she developed slowly progressive but marked hemiparesis of the right limbs and slight neurocognitive impairment. At the age of 71, she needs a cane to walk, but seizures are controlled without AEDs.
 Hematoxylin–eosin staining with perivascular and diffuse lymphocyte infiltration and a microglial nodule. (B) CD8 immunohistochemistry with marked perivascular and parenchymal CD8+ T lymphocytes. (C) NeuN immunohistochemistry with patchy areas of cortical neuron loss.")
Brain histopathological examination of patient 2. Brain biopsy histopathological findings. (A) Hematoxylin–eosin staining with perivascular and diffuse lymphocyte infiltration and a microglial nodule. (B) CD8 immunohistochemistry with marked perivascular and parenchymal CD8+ T lymphocytes. (C) NeuN immunohistochemistry with patchy areas of cortical neuron loss.
![Complementary studies of patient 2. (A) EEG (2020). Bipolar circular montage showing normal background activity, with slow spike and wave paroxysms collected in the temporal region of the left hemisphere. (B) Brain 18F-FDG PET (2022). Left frontal, striatal, and thalamic hypometabolism. (C) Brain MRI (2019). Axial FLAIR-weighted image and coronal FLAIR-weighted image. Left frontal parasagittal lesion without gadolinium enhancement or mass effect, and very mild left hemispheric atrophy. (D) Brain MRI (2020). Axial and coronal FLAIR-weighted images showing significant growth of the lesion over the left frontal lobe, parietal lobe, and periventricular area, without gadolinium enhancement or mass effect, as well as notable ipsilateral atrophy. 18F-FDG PET: positron emission tomography with 2-deoxy-2-[fluorine-18] fluoro-d-glucose; EEG: electroencephalography study; FLAIR: fluid-attenuated inversion recovery; MRI: magnetic resonance imaging.](https://static.elsevier.es/multimedia/02134853/unassign/S0213485324001130/v1_202409020413/en/main.assets/gr4.jpeg?xkr=ue/ImdikoIMrsJoerZ+w997EogCnBdOOD93cPFbanNcnxJVYM2BEAEuZ5XZLhi9i85vcbshWVQPfl+0x3NpqwiqkNtigdeFuE9Zyvaz/gsKNfR4ysGK+modMU8s6xaRFTe8NUh3LUk+0JlqzujjzTngRDBXjzKcXNFsxhRyIRupVXqhA2WKaQwENP5ihuSTdMsXYtYPqROenHt8e0adXU42kSRsOrOcUsOtKYTko1sXo1cpyAR87egsyQQA1gPXqX/qWCNcoLYoA1RChn4l18zEw4MdWiMSYGxNAItvBfBk= "Complementary studies of patient 2. (A) EEG (2020). Bipolar circular montage showing normal background activity, with slow spike and wave paroxysms collected in the temporal region of the left hemisphere. (B) Brain 18F-FDG PET (2022). Left frontal, striatal, and thalamic hypometabolism. (C) Brain MRI (2019). Axial FLAIR-weighted image and coronal FLAIR-weighted image. Left frontal parasagittal lesion without gadolinium enhancement or mass effect, and very mild left hemispheric atrophy. (D) Brain MRI (2020). Axial and coronal FLAIR-weighted images showing significant growth of the lesion over the left frontal lobe, parietal lobe, and periventricular area, without gadolinium enhancement or mass effect, as well as notable ipsilateral atrophy. 18F-FDG PET: positron emission tomography with 2-deoxy-2-[fluorine-18] fluoro-d-glucose; EEG: electroencephalography study; FLAIR: fluid-attenuated inversion recovery; MRI: magnetic resonance imaging.")
Complementary studies of patient 2. (A) EEG (2020). Bipolar circular montage showing normal background activity, with slow spike and wave paroxysms collected in the temporal region of the left hemisphere. (B) Brain 18F-FDG PET (2022). Left frontal, striatal, and thalamic hypometabolism. (C) Brain MRI (2019). Axial FLAIR-weighted image and coronal FLAIR-weighted image. Left frontal parasagittal lesion without gadolinium enhancement or mass effect, and very mild left hemispheric atrophy. (D) Brain MRI (2020). Axial and coronal FLAIR-weighted images showing significant growth of the lesion over the left frontal lobe, parietal lobe, and periventricular area, without gadolinium enhancement or mass effect, as well as notable ipsilateral atrophy. 18F-FDG PET: positron emission tomography with 2-deoxy-2-[fluorine-18] fluoro-d-glucose; EEG: electroencephalography study; FLAIR: fluid-attenuated inversion recovery; MRI: magnetic resonance imaging.
A 23-year-old woman without relevant clinical history presented 4 years’ history of involuntary movements in the left lower limb, triggered by activity. Initial complementary test findings were normal. Several AEDs were trialed, without any improvement. Subsequently, a study revealed giant somatosensory evoked potentials during stimulation of the left tibial nerve, suggestive of cortical hyperexcitability. A year later, involuntary movements were present even at rest, and were exacerbated with tactile stimuli. She also presented generalized tonic–clonic seizures. AED therapy was adjusted, which controlled the seizures but not the continuous movements of the foot, which were compatible with epilepsia partialis continua (EPC). Brain MRI showed frontobasal hyperintensity and cortical atrophy. Autoimmunity and genetic progressive myoclonic epilepsy were ruled out. A video-EEG study was carried out, supporting the diagnosis (Video 1). Brain positron emission tomography with 2-deoxy-2-[fluorine-18] fluoro-d-glucose (18F-FDG PET), MRI arterial-spin-labeling (ASL), and neuropsychological studies were also performed (Table 1, Fig. 5). Under the diagnosis of LORE, first-line immunotherapy was started but was not effective; subsequently, adalimumab was initiated, achieving a relevant clinical improvement in terms of EPC severity and seizure control. After a year, the patient remained stable, with slight progression of the disease.
![Complementary studies of patient 3. (A) EEG (2019). Referential average montage showing normal background activity with superimposition of occasional right frontal spike-and-wave paroxysms, with slight diffusion to contralateral homologous regions. (B) Video-EEG (2021). Bipolar circular montage showing normal background activity with superimposition of frequent spike and spike-and-wave paroxysms in the right fronto-parietal area (C4, Fp2, F8), with great tendency to diffusion to the central area; occasional periodic presentation. These paroxysms most often appeared in phase N2<N3<REM. Arrhythmic focal myoclonus with no EEG correlation. (C) 18F-FDG PET, co-registered with brain MRI. Axial image. Hypometabolic foci in the right frontal lobe; the extent and severity of hypometabolism may be overestimated due to a seizure that occurred before the PET-CT study was conducted. The right cerebellar hemisphere presents hypometabolism. (D) Brain MRI (2022). The image shows 2 T2-hyperintense lesions in the basal region of the right frontal lobe, associated with discrete cortical atrophy and juxtacortical gliosis. (E) Brain ASL MRI (2022). Right frontal hypoperfusion. 18F-FDG PET: positron emission tomography with 2-deoxy-2-[fluorine-18] fluoro-d-glucose; ASL: arterial spin labeling; EEG: electroencephalography study; FLAIR: fluid-attenuated inversion recovery; MRI: magnetic resonance imaging.](https://static.elsevier.es/multimedia/02134853/unassign/S0213485324001130/v1_202409020413/en/main.assets/gr5.jpeg?xkr=ue/ImdikoIMrsJoerZ+w997EogCnBdOOD93cPFbanNcnxJVYM2BEAEuZ5XZLhi9i85vcbshWVQPfl+0x3NpqwiqkNtigdeFuE9Zyvaz/gsKNfR4ysGK+modMU8s6xaRFTe8NUh3LUk+0JlqzujjzTngRDBXjzKcXNFsxhRyIRupVXqhA2WKaQwENP5ihuSTdMsXYtYPqROenHt8e0adXU42kSRsOrOcUsOtKYTko1sXo1cpyAR87egsyQQA1gPXqX/qWCNcoLYoA1RChn4l18zEw4MdWiMSYGxNAItvBfBk= "Complementary studies of patient 3. (A) EEG (2019). Referential average montage showing normal background activity with superimposition of occasional right frontal spike-and-wave paroxysms, with slight diffusion to contralateral homologous regions. (B) Video-EEG (2021). Bipolar circular montage showing normal background activity with superimposition of frequent spike and spike-and-wave paroxysms in the right fronto-parietal area (C4, Fp2, F8), with great tendency to diffusion to the central area; occasional periodic presentation. These paroxysms most often appeared in phase N2<N3<REM. Arrhythmic focal myoclonus with no EEG correlation. (C) 18F-FDG PET, co-registered with brain MRI. Axial image. Hypometabolic foci in the right frontal lobe; the extent and severity of hypometabolism may be overestimated due to a seizure that occurred before the PET-CT study was conducted. The right cerebellar hemisphere presents hypometabolism. (D) Brain MRI (2022). The image shows 2 T2-hyperintense lesions in the basal region of the right frontal lobe, associated with discrete cortical atrophy and juxtacortical gliosis. (E) Brain ASL MRI (2022). Right frontal hypoperfusion. 18F-FDG PET: positron emission tomography with 2-deoxy-2-[fluorine-18] fluoro-d-glucose; ASL: arterial spin labeling; EEG: electroencephalography study; FLAIR: fluid-attenuated inversion recovery; MRI: magnetic resonance imaging.")
Complementary studies of patient 3. (A) EEG (2019). Referential average montage showing normal background activity with superimposition of occasional right frontal spike-and-wave paroxysms, with slight diffusion to contralateral homologous regions. (B) Video-EEG (2021). Bipolar circular montage showing normal background activity with superimposition of frequent spike and spike-and-wave paroxysms in the right fronto-parietal area (C4, Fp2, F8), with great tendency to diffusion to the central area; occasional periodic presentation. These paroxysms most often appeared in phase N2<N3<REM. Arrhythmic focal myoclonus with no EEG correlation. (C) 18F-FDG PET, co-registered with brain MRI. Axial image. Hypometabolic foci in the right frontal lobe; the extent and severity of hypometabolism may be overestimated due to a seizure that occurred before the PET-CT study was conducted. The right cerebellar hemisphere presents hypometabolism. (D) Brain MRI (2022). The image shows 2 T2-hyperintense lesions in the basal region of the right frontal lobe, associated with discrete cortical atrophy and juxtacortical gliosis. (E) Brain ASL MRI (2022). Right frontal hypoperfusion. 18F-FDG PET: positron emission tomography with 2-deoxy-2-[fluorine-18] fluoro-d-glucose; ASL: arterial spin labeling; EEG: electroencephalography study; FLAIR: fluid-attenuated inversion recovery; MRI: magnetic resonance imaging.
The typical childhood-onset form usually starts at 6–7 years of age, and 3 clinical stages may be distinguished: firstly, a prodromal stage with mild hemiparesis or infrequent seizures, with a median duration of 7 months; secondly, an acute stage defined by frequent focal seizures and progressive neurological and neurocognitive impairment, with a median duration of 8 months; and finally, a residual stage, with severe fixed motor and cognitive deficits and refractory epilepsy, which is usually established between the first and the third year after onset.2,3,5,21
However, few data are available regarding patients with LORE. Their description has been based mainly on case reports and a few case series, and precise characterization of the disease remains subject to debate. Late onset is frequently defined as presentation after 12 years of age,4,7 but onset is highly variable, with a mean (standard deviation) age of 24 (12) years (range, 12–62).7,22 The prodromal stage may precede the acute stage by several years, making early diagnosis a challenge. In addition, the acute and residual stages present slower and milder progression, with delayed and less frequent occurrence of EPC, neurological deficits, focal cortical atrophy, and cognitive impairment, and therefore, an overall better prognosis.7,23,24
The European consensus diagnostic criteria for RE3 (Table 2) were proposed in 2005, but were intended for pediatric patients, so their application in adult cases may be complex. Strikingly, at least 67% of reported patients with LORE were found to fulfill these criteria, based on Part B in 77% of cases (as in our patient 2), requiring prolonged follow-up, and part A in 23% (as in our patients 1 and 3), enabling earlier diagnosis.7
European consensus diagnostic criteria for Rasmussen encephalitis.
| Part A: | |
| 1. Clinical | Focal seizures (with or without EPC) and unilateral cortical deficit(s) |
| 2. EEG | Unihemispheric slowing with/without epileptiform activity and unilateral seizure onset |
| 3. MRI | Unihemispheric focal cortical atrophy and at least one of the following:• Gray or white matter T2/FLAIR hyperintense signal• Hyperintense signal or atrophy of the ipsilateral caudate head |
| Part B: | |
| 1. Clinical | EPC or progressivea unilateral cortical deficit(s) |
| 2. MRI | Progressivea unihemispheric focal cortical atrophy |
| 3. Histopathology | T cell-dominated encephalitis with activated microglial cells (typically, but not necessarily, forming nodules) and reactive astrogliosis.Numerous parenchymal macrophages, B cells or plasma cells, or viral inclusion bodies exclude the diagnosis of RE. |
RE can be diagnosed if either all 3 Part A criteria or 2 out of 3 Part B criteria are present.
“Progressive” means that at least 2 sequential clinical examinations or MRI studies are required to meet the respective criteria. To indicate clinical progression, each of these examinations must document a neurological deficit, and this must increase over time. To indicate progressive hemiatrophy, each of these MRI studies must show hemiatrophy, and this must increase over time.
EEG: electroencephalography study; EPC: epilepsia partialis continua; FLAIR: fluid attenuated inversion recovery sequence; MRI: magnetic resonance imaging; RE: Rasmussen encephalitis.
The most frequent presentation is focal epilepsy. Classically, unihemispheric and polymorphic seizures, EPC, and refractoriness have been described as the main features of epilepsy in patients with RE.7
Epilepsy, especially motor seizures, may be the only clinical manifestation at onset. Focal aware seizures, mainly the motor type, are most frequent, followed by focal to bilateral tonic–clonic seizures in both children and adults.2,3,5,21,23,24 However, other types of seizures may appear. For example, patient 1 suffered from sensory visual seizures, related to occipital lobe involvement.
EPC is reported to occur during the disease course in 56–92% of pediatric patients,5,21,25 as compared to 31% of adult patients.7 EPC was first described by Kozhevnikov,26 and may be the initial presentation in LORE,24,27 as was the case in patient 3.
Interestingly, some patients develop seizures only several months or even years after the clinical onset,28 as in patient 2.
Neurocognitive and neurological impairmentNeurocognitive decline often presents slower and less severe progression, with a prolonged delay of several years after the first seizure. These alterations may present in fewer than half of patients during follow-up.7,29 Neuropsychological deficits involve alterations in such domains as attention, verbal memory, speech fluency, abstraction, and learning.24 Our patients had mainly frontal symptoms as well as auditory memory impairment, consistent with MRI findings. Patients 1 and 2 presented mild cognitive impairment after 12 and 4 years of follow-up, respectively. Additionally, psychiatric symptoms may be present or may even be the most prominent feature.30 These patients showed mild or slight depression but no other psychiatric symptoms.
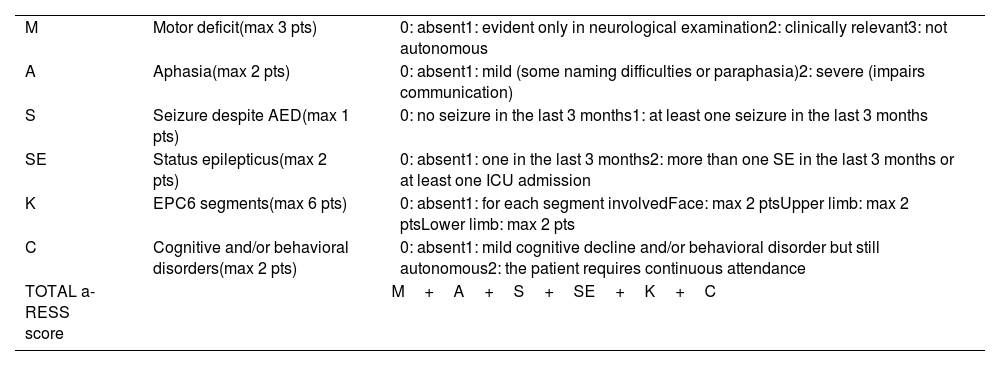
Regarding neurological deficits in LORE, the most common symptoms are hemiparesis and aphasia if the dominant hemisphere is affected, occurring in 95% of patients after a mean follow-up time of 3 years after seizure onset. Despite this, atypical cases have been described in association with bilateral31 or brainstem involvement,32 as well as a possible overlap with Parry-Romberg syndrome.33 The clinical monitoring of these patients may be challenging. Therefore, the Adult-onset Rasmussen Encephalitis Severity Scale (a-RESS) has been proposed to assess the overall disease severity (Table 3).34 Our patients reached a maximum score of 4, with a slight decrease (to 3 points) at the cut-off due to an improvement in seizure frequency or EPC intensity.
The a-RESS scale.
| M | Motor deficit(max 3 pts) | 0: absent1: evident only in neurological examination2: clinically relevant3: not autonomous |
| A | Aphasia(max 2 pts) | 0: absent1: mild (some naming difficulties or paraphasia)2: severe (impairs communication) |
| S | Seizure despite AED(max 1 pts) | 0: no seizure in the last 3 months1: at least one seizure in the last 3 months |
| SE | Status epilepticus(max 2 pts) | 0: absent1: one in the last 3 months2: more than one SE in the last 3 months or at least one ICU admission |
| K | EPC6 segments(max 6 pts) | 0: absent1: for each segment involvedFace: max 2 ptsUpper limb: max 2 ptsLower limb: max 2 pts |
| C | Cognitive and/or behavioral disorders(max 2 pts) | 0: absent1: mild cognitive decline and/or behavioral disorder but still autonomous2: the patient requires continuous attendance |
| TOTAL a-RESS score | M+A+S+SE+K+C | |
a-RESS: Adult-onset Rasmussen Encephalitis Severity Scale; AED: antiepileptic drug; EPC: epilepsia partialis continua; ICU: intensive care unit; SE: status epilepticus.
An association between RE and serum or CSF autoimmune markers7 or autoimmune diseases has been reported. Hashimoto thyroiditis, ulcerative colitis, Crohn disease, SLE, and bilateral uveitis have been described as possible comorbidities,35–37 suggesting a complex autoimmune background in patients with LORE. For instance, patient 2 developed a cutaneous lesion compatible with SLE 4 years after clinical onset.
Movement disordersMovement disorders, such as dystonia or chorea, may also be present.38–40 In fact, parkinsonism and even corticobasal syndrome have been described as atypical symptoms in RE.41
Complementary testingDespite research efforts, no clear serological or CSF biomarkers have been discovered to date. Some non-specific serum and CSF findings may be observed, such as an increased CSF protein level or/and increased cell counts, or non-specific presence of oligoclonal bands. Moreover, the GluR3 antibodies are detected in the CSF of 6% of patients with LORE.7
Patient 1 tested positive for SOX-1, an antibody previously considered a biomarker of paraneoplastic neurological disorders associated with lung tumors.42 In this case, there was no evidence of an underlying tumor during follow-up. Similarly, patient 2 presented low GAD antibody titers. We hypothesize that some “autoimmune overlap” may be present in these patients, without a specific clinical correlation. However, patient 2 tested positive for ANA antibodies, and after 4 years she developed clinical signs of SLE, which may have arisen later as she was being treated with azathioprine.
EEG is a useful study whose findings progressively become abnormal over time, frequently showing impaired background activity with focal slow waves or epileptic abnormalities,3,7,24,25 which sometimes progress to an extra-rolandic network in the advanced phase of the disease,43 or spread to the contralateral hemisphere in rare cases.23 Our patients’ EEG studies showed epileptic discharges and unihemispheric focal slowing in all patients except for patient 3, who had EPC but normal background activity. If no surface EEG changes are detected, subdural electrodes may be of value.44
In addition, somatosensory evoked potentials recorded over the affected hemisphere may be abnormally increased (>10μV), as observed in patient 3.24
Regarding brain MRI, unihemispheric atrophy and involvement of the head of the caudate constitute a major finding. Among our patients, atrophy most frequently involved the frontal lobe. Previous evidence indicates posterior predilection in LORE,2,6 whereas a recent publication, including 13 adult patients, concluded that frontal and temporo-insular regions were the most frequently affected areas. Nevertheless, significant involvement of the parietal and occipital lobes was also present, mostly during advanced stages of disease progression.29 In this regard, more prospective studies are needed before any conclusions may be drawn.
Another prominent finding is T2WI and FLAIR hyperintensities involving the gray and white matter45,46; and dynamic changes may occur, depending on the stage of the disease.47 Both atrophic and hyperintense alterations have a long-lasting course, with a progression from mild to severe, even over extremely long periods (decades), regardless of surgical or medical therapy.46
Other brain MRI findings, such as chronic ischemic changes or cortical dysplasia, have also been described,48,49 suggesting that a dual or a triple pathology might be present in these patients.
Functional neuroimaging, especially brain 18F-FDG PET, may provide valuable biomarkers, most frequently showing extensive unihemispheric hypometabolism correlating with MRI findings,46,50 as observed in our patients. In addition, it seems that patients with focal cortical myoclonus, such as patient 3, may present milder atrophy on MRI. Despite this, extensive metabolic impairment is shown in brain 18F-FDG PET, suggesting that it may contribute significantly to early diagnosis of LORE, particularly in the myoclonic subgroup.24
Similarly, cerebral blood flow single photon emission computed tomography (SPECT) usually shows changes over the affected hemisphere, with hypoperfusion being the most prevalent.51–53 Other modalities, such as functional MRI,54 can also be useful. Patient 3 had congruent findings, showing right frontal hypoperfusion in the ASL study.
To summarize, both MRI and functional neuroimaging results showed an adequate correlation with the patients’ symptoms.
TreatmentThe aim of treatment is to control both seizures and disease progression. However, the best treatment strategy for LORE is still unclear.
Antiepileptic therapyAED therapy remains a treatment for the symptomatic control of seizures. These drugs seem useful for decreasing the risk of focal to bilateral tonic–clonic seizures, with less reassuring results in EPC.19,55 Despite the lack of evidence that any particular AED is superior in LORE, a recent publication ascertained that sodium channel blockers may be superior to other AEDs in autoimmune-related focal epilepsy.56
Epilepsy surgeryClassically, the only established treatment to control seizures and to stop neurological deterioration in pediatric patients was surgery. Surgical techniques include complete hemispheric resection or hemispheric disconnection (HD). The rate of success in terms of seizure freedom is 80%, although the treatment entails a loss of fine motor functions in the contralateral hand, spastic hemiplegia, homonymous hemianopsia, and a form of expressive aphasia in patients undergoing left-sided HD.3 Therefore, these options may not be feasible in adult patients, whose brain plasticity and subsequent recovery may be limited.24 In these cases, focal resection is the most widely used technique, with good results in terms of seizure freedom and safety, although its efficacy in terms of disease progression remains unclear. This conservative surgical option is recommended in highly selected clinical settings, and patient-tailored surgery approaches may be considered.57,58 Current evidence strongly suggests HD as the treatment of choice for the most aggressive forms in children,59,60 although decision-making regarding surgery in LORE involving the dominant hemisphere, slowly progressive forms, or limited neurological deficits remains a challenge. In this context, some studies have reported the efficacy of other surgical treatments in selected cases, such as neurostimulation with vagus nerve stimulators,61 brain-responsive stimulation,62 unilateral pallidal stimulation for symptomatic dystonia,63 or transcranial direct current stimulation.64
ImmunotherapyImmunotherapy represents a promising new therapeutic approach in different circumstances. Proposed therapeutic algorithms have recently been updated.3,19,55,65
- •
First-line treatment
First-line treatments include intravenous immunoglobulins (IVIG), plasmapheresis, and steroid therapy. These have a rapid but mostly transient effect and may be used either as a first-line treatment followed by another immunotherapy, or in the event of rapid worsening of the disease (e.g., status epilepticus).3,19,55,65 In LORE, these immunotherapies achieved a good overall response in both epilepsy and neurological deterioration, with positive responses in 61% of patients,7 similar to the results in our series.
According to the available evidence, high-dose corticosteroids are recommended as a first-line treatment due to their effect on cellular immunity. Despite being an effective treatment,25,55,66,67 they should be followed by another steroid-sparing immunotherapy to limit their long-term side effects.65
IVIG has been proposed as additional first-line or add-on therapy, acting on both humoral and cellular immunity. The overall efficacy of IVIG in reducing seizures is 30%, and appears to be lower than that of corticosteroids.19 However, patients may respond remarkably in terms of seizure frequency and EPC resolution,68,69 with fewer side effects than those associated with high-dose corticosteroids.65
Plasmapheresis reduces levels of autoantibodies, other immune mediators, and complement factors. Currently, the evidence regarding the efficacy of plasmapheresis for LORE remains unclear. Overall, plasmapheresis has low response rates with frequent relapses in long-term follow-up, but it might be useful when other first-line treatments fail or before rapid switch to another long-term immunotherapy.19,70
Patient 1 was treated with IVIG. Due to the good tolerance and slow progression, second-line treatment was ultimately not initiated. However, after 12 years of follow-up the patient developed moderate neurocognitive and motor decline. It is unclear whether the clinical situation would differ if a second-line treatment had been started earlier.
- •
Second-line treatment
Second-line treatment remains underexplored. These treatments have a longer duration of action with a better safety profile.19
Immunosuppressive agentsThe best evidence includes tacrolimus, a calcineurin inhibitor that acts by blocking IL-2 transcription, leading to a reduction of T-cell activity with potential to affect the underlying progression of the disease. However, it has shown low effectiveness in treating seizures.4,25,71 When compared with IVIG, tacrolimus was associated with comparable clinical response, but a higher rate of adverse events.4 The use of tacrolimus seems limited in clinical practice.19
Another noteworthy treatment is azathioprine, an inhibitor of purine synthesis that results in decreased T-cell proliferation and apoptosis of activated T-cells.72,73 Patient 2 was receiving this treatment, which has been shown to have good efficacy for seizure control but a small impact on atrophy and disability progression.19 It was chosen because of the wide-ranging experience reported in other immune-mediated neurological conditions. After 4 years of follow-up the patient was seizure-free but developed a marked motor decline.
Mycophenolate mofetil is another alternative that inhibits purine synthesis in lymphocytes, inducing apoptosis of activated T lymphocytes and reducing the recruitment of lymphocytes, although there is scarce and controversial evidence regarding its use in LORE.19,55
Biologic drugsAmong biologic agents, rituximab is an anti-CD20 monoclonal antibody that has been used in a small number of published case reports, with variable efficacy.74,75 Rituximab has recently shown a significant benefit in reducing seizure burden, as well as halting the progression of neurological deficits.76,77
Natalizumab, an antibody targeting integrin alpha-4, alters the ability of T lymphocytes to cross the blood-brain barrier and has been described in isolated case reports, showing good clinical efficacy.78
More recently, adalimumab is a fully humanized monoclonal antibody targeting TNF-α, which has arisen as a promising new therapeutic option. It was reported to reduce seizure frequency by more than 50% in 5/11 patients, 3 of whom presented stabilization of their neurological decline.79 A trial of the drug is currently underway (code: NCT04003922). Patient 3 did not present any improvement in EPC with first-line therapy and AEDs, so a second-line treatment was initiated. Adalimumab was chosen due to its promising results in terms of efficacy and safety.
Anakinra is a recombinant and slightly modified-form interleukin-1 (IL-1) receptor antagonist (IL-1Ra) that blocks the inflammatory activity of both IL-1α and IL-1β.19 It has been reported in a single case as a successful treatment, with complete cessation of seizures for a period of 13 months without recurrence.80
Finally, the emergence of molecular diagnostic techniques, such as cytokine expression profile analysis, will provide us with the information to perform precision medicine with individualized and targeted therapies.
Conclusions- •
LORE is associated with better prognosis and less severe neurological decline than RE in pediatric patients.
- •
The most frequent clinical presentation is focal epilepsy, but RE may present with other atypical symptoms at onset.
- •
Diagnosis is often delayed due to mild severity at onset and slower progression.
- •
Future studies should assess the diagnostic value of brain 18F-FDG PET/CT biomarkers.
- •
Treatment must be patient-tailored, but immunotherapy has proven to be a useful treatment in different scenarios.
- •
Corticosteroids appear to be the most effective first-line therapy, and can be supplemented with IVIG if necessary.
- •
Among the second-line treatments, azathioprine, adalimumab, and rituximab seem to have the most robust evidence in terms of seizure control and disease progression.
- •
Further research with prospective and comparative studies is necessary to define the distinctive features of these patients and to clarify the best treatment strategy.
The current study was conducted in accordance with the ethical code of the World Health Organization. Informed consent was obtained from the patients.
Study fundingsThis research has not received specific aid from any public, private, or non-profit entity.
Conflicts of interestWe have no conflicts of interest to declare.
This study was made possible thanks to a grant from the Ad-hoc Committee for Young Neurologists of the SEN and the Spanish Society of Neurology.

