




The complex process of amyloid-β (Aβ) transportation across the blood–brain and blood–cerebrospinal fluid barriers is crucial for preventing Aβ accumulation, which linked to dementia and neurodegeneration. This review explores therapeutic plasma exchange with albumin replacement in Alzheimer's disease, based on the dynamics of amyloid-β between the brain, plasma, and cerebrospinal fluid.
MethodologyA comprehensive literature review was conducted using PubMed/Medline, Cochrane Library, and open databases (bioRxiv, MedRixv, preprint.org) up to April 30, 2023. The first search utilized the following MeSH terms and keywords: ‘Plasma Exchange’, ‘Plasmapheresis’, ‘Therapeutic plasma exchange’, ‘Apheresis’, ‘Aβ’, ‘p-tau’, ‘Total-tau’, ‘Alzheimer's disease’, ‘Cognitive dysfunction’, ‘neurodegenerative diseases’, ‘centrifugation’, ‘membranous’, and ‘filtration’ in the Title/Abstract, yielding 146 results. A second search with the keywords: ‘Albumin’, ‘Aβ’, ‘BBB’, ‘Alzheimer's dementia’, and ‘Nerve degeneration’ resulted in 125 additional articles for analysis. Finally, a third search using keywords: ‘Albumin structural domains’, ‘Albumin-Aβ interactions’, ‘Albumin-endothelial interactions’, and ‘Post-Translational Modification’ produced 193 results for further review.
Results/DiscussionTherapeutic plasma exchange shows potential as a disease-modifying therapy for dementia, specifically for Alzheimer's disease. Additionally, the promising role of albumin supplementation in cognitive improvement has attracted attention. However, clinical evidence supporting therapeutic plasma exchange for dementia remains limited, necessitating further research and development to mitigate potential adverse effects. A deeper understanding of the molecular dynamics of Aβ transportation and the mechanisms of therapeutic plasma exchange is essential. A critical evaluation of existing evidence highlights the importance of balancing potential benefits with associated risks, which will guide the development and application of these treatments in neurodegenerative diseases.
El complejo proceso de transporte del amiloide-β (Aβ) a través de las barreras sangre-cerebro y sangre-líquido cefalorraquídeo es crucial para prevenir la acumulación de Aβ, vinculada a la demencia y la neurodegeneración. Esta revisión explora el intercambio terapéutico de plasma con reemplazo de albúmina en la enfermedad de Alzheimer, fundamentado en la dinámica del Aβ entre el cerebro, el plasma y el líquido cefalorraquídeo.
MetodologíaSe realizó una revisión exhaustiva de la literatura utilizando PubMed/Medline, Cochrane Library y bases de datos abiertas (bioRxiv, MedRixv, preprint.org) hasta el 30 de abril de 2023. La primera búsqueda utilizó los siguientes términos MeSH y palabras clave: ‘Plasma Exchange’, ‘Plasmapheresis’, ‘Therapeutic plasma exchange’, ‘Apheresis’, ‘Aβ’, ‘p-tau’, ‘Total-tau’, ‘Alzheimer's disease’, ‘Cognitive dysfunction’, ‘neurodegenerative diseases’, ‘centrifugation’, ‘membranous’ y ‘filtration’ en el Título/Abstract, obteniendo 146 resultados. Una segunda búsqueda con las palabras clave: ‘Albumin’, ‘Aβ’, ‘BBB’, ‘Alzheimer's dementia’ y ‘Nerve degeneration’ resultó en 125 artículos adicionales para análisis. Finalmente, una tercera búsqueda utilizando las palabras clave: ‘Albumin structural domains’, ‘Albumin-Aβ interactions’, ‘Albumin-endothelial interactions’ y ‘Post-Translational Modification’ produjo 193 resultados para una revisión adicional.
Resultados y discusiónEl intercambio terapéutico de plasma muestra potencial como una terapia modificadora de la enfermedad para la demencia, específicamente para la enfermedad de Alzheimer. Además, el papel prometedor de la suplementación con albúmina en la mejora cognitiva ha llamado la atención. Sin embargo, la evidencia clínica que respalda el intercambio terapéutico de plasma para la demencia sigue siendo limitada, lo que requiere más investigación y desarrollo para mitigar posibles efectos adversos. Es esencial una comprensión más profunda de la dinámica molecular del transporte de Aβ y los mecanismos del intercambio terapéutico de plasma. Una evaluación crítica de la evidencia existente resalta la importancia de equilibrar los beneficios potenciales con los riesgos asociados, lo cual guiará el desarrollo y la aplicación de estos tratamientos en enfermedades neurodegenerativas.
Alzheimer's disease (AD) encompasses cognitive deficits and behavioral disturbances that ultimately lead to dementia and the cortical accumulation of pathological lesions.1 Despite being the sixth leading cause of death in the USA and the most prevalent form of dementia, AD has no cure, and its global burden on healthcare costs is enormous.2 Currently, approximately 57.4million people are living with AD and related dementias. The disease has risen from being the 41st largest cause of disability-adjusted life years during 1990 to the 23rd in 2016, with an estimated 150 million cases projected globally by 2050.3,4 Therefore, a global effort is needed to explore treatments beyond the current guidelines, which primarily address symptomatic management rather than the underlying neuronal damage associated with morbidity and mortality.
Noteworthy to mention, except for a few genetic aberrations in the forms of autosomal dominant mutations related to proteolytic modification of amyloid precursor protein (APP), the etio-pathological basis of the majority of “sporadic AD” is a gray area.5 For that instance, it is also imperative to note that the widely accepted theory supporting the connection between ‘amyloid-β and AD’ is being challenged, and the implication of amyloid-β (Aβ) remains an enigma. Apart from the predominantly accepted role of Aβ, total Tau, and phosphorylated tau (p-Tau), recent findings on the neuroinflammatory association as a significant driver and accelerator in AD pathogenesis from early cognitive impairment to the late stage has opened up a new dimension in the therapeutic intervention.6,7 On this note, the role of therapeutic plasma exchange in neurological disorders, including auto-immune demyelinating disorders, paraneoplastic neurological syndromes, and neuronal surface autoantibodies-associated syndromes has been proven tremendously efficacious.8 The efficacy of therapeutic plasma exchange depends on the fractional percentage of plasma removed, distribution, and production of the pathological substance alongside its equilibrium between plasma and organs.9 Besides, the role of critical endogenous factors such as serum and cerebrospinal fluid (CSF)-albumin in AD behind maintaining such inter-compartmental equilibrium of pathological substances like Aβ and inflammatory complexes needs a more profound understanding.10,11
This review explores therapeutic plasma exchange with albumin replacement in AD, based on the dynamics of Aβ between the brain, plasma, and CSF.
MethodsSearch strategy for the literature reviewA comprehensive literature review was conducted using PubMed/Medline, Cochrane Library, and open databases (bioRxiv, MedRixv, preprint.org) up to April 30, 2023. The first search utilized the following MeSH terms and keywords: ‘Plasma Exchange’, ‘Plasmapheresis’, ‘Therapeutic plasma exchange’, ‘Apheresis’, ‘Aβ’, ‘p-tau’, ‘Total-tau’, ‘Alzheimer's disease’, ‘Cognitive dysfunction’, ‘neurodegenerative diseases’, ‘centrifugation’, ‘membranous’, and ‘filtration’ in the Title/Abstract, yielding 146 results. A second search with the keywords: ‘Albumin’, ‘Aβ’, ‘BBB’, ‘Alzheimer's dementia’, and ‘Nerve degeneration’ resulted in 125 additional articles for analysis. Finally, a third search using keywords: ‘Albumin structural domains’, ‘Albumin-Aβ interactions’, ‘Albumin-endothelial interactions’, and ‘Post-Translational Modification’ produced 193 results for further review. Three researchers (SD, GS, and VS) completed the literature search. RM, DL, and SSD scrutinized the abstracts obtained. RM, SSD, and JBL analyzed the full texts in case the abstracts were relevant. For articles without abstracts or with less detailed abstracts, SD directly analyzed the full text. Once the finding was compiled, they were reviewed by RM, SD, VS, GS, DL, SSD, and JBL.
Results/discussion- 1.
The dynamic relationship between serum, brain parenchyma, and CSF-Aβ load: The theory of ‘peripheral sink’ and therapeutic plasma exchange
Familial AD is primarily associated with increased Aβ1–42 formation, while sporadic AD is likely due to a long-term imbalance between different forms of Aβ peptide production and clearance. Physiologically, interstitial Aβ monomers are in equilibrium with Aβ-oligomers.12 Larger fibrils are cleared off the brain parenchyma via metabolic degradation and transportation across the blood–brain barrier (BBB) into the plasma and perivascular space and via the CSF system.12,13 The most accepted hypothesis in Aβ-mediated brain injury in AD is the ‘amyloid cascade’, comprising of amyloid accumulation in brain tissues, formation of toxic oligomeric and intermediate form of Aβ, amyloid plaques, inflammation, and the formation of neurofibrillary tangles.14–16 The theory of the ‘peripheral sink’ of Aβ consisting of plasma Aβ sink and CSF-Aβ sink, tries to explain Aβ-accumulation due to deranged homeostasis between its formation and elimination.17
In order to traverse BBB, soluble Aβ from the extracellular tissue space needs to reach brain endothelial cells after crossing the capillary basement membrane, pericytes, and astrocytic endfeet.18–20 Several studies have reflected significant alteration of Aβ transportation across BBB with aging due to reduced Aβ-flux and alteration in Aβ transporters.21,22 The blood–CSF barrier (BCSFB) also undergoes age-related changes, adding to the Aβ burden.23,24 Hence, it is crucial to understand this three-point homeostatic relationship between brain parenchymal-Aβ production, plasma-Aβ, and CSF-Aβ load. In this section, we aim to describe Aβ transportation across BBB and BCSFB in physiological and diseased conditions and the role of human serum albumin (HSA) in maintaining this tight homeostasis.
- 1.1
Transportation of Aβ across BBB: an insight into ‘Plasma-Aβ sink.’
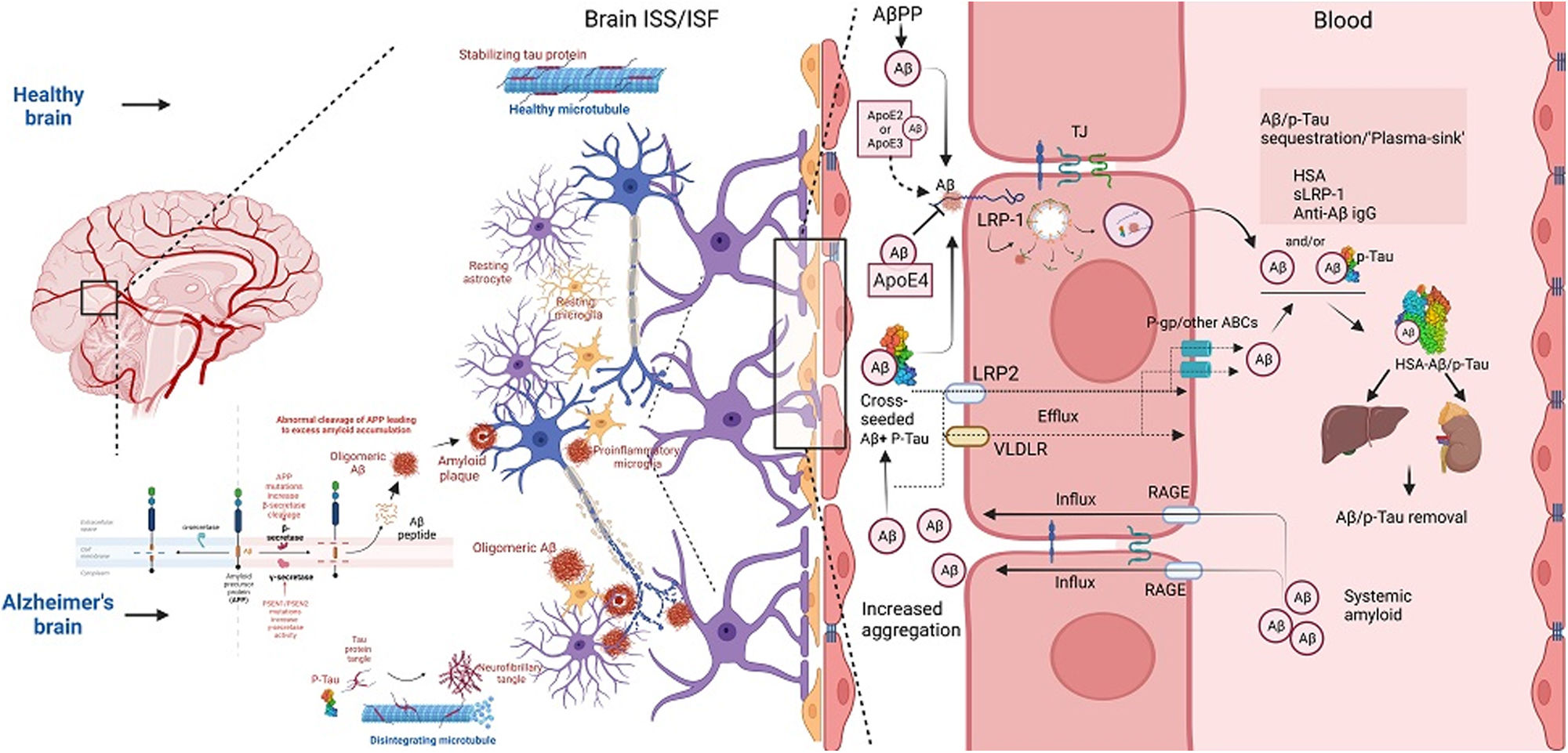
Aβ transportation across the BBB is a highly regulated and complicated multi-step efflux and influx process.25,26 Although the detailed picture of the relationship between plasma and interstitial fluid (ISF) is still poorly understood, changes in its properties have been extensively reported in cases of abnormal Aβ accumulation in the brain's interstitial space microarchitecture27 (Fig. 1). In AD, the Aβ transporter low-density lipoprotein receptor-related protein (LRP) 1 (LRP-1) on the abluminal side of capillary endothelium is decreased, while the receptor for advanced glycation end products (RAGE) on the luminal and microvascular endothelium is significantly increased.28 The relative alteration of these transporters is positively associated with net interstitial soluble Aβ accumulation, leading to insoluble and toxic Aβ intermediate formation.24,26 Hypercholesterolemia, positively correlated with spatial learning and memory impairment, can trigger alterations leading to cerebral and brain microvascular endothelial Aβ accumulation.29 In addition, peripherally produced Aβ by various cell types is transported into the brain across the BBB through transcytosis facilitated by receptors such as RAGE.28 Notably, Aβ-RAGE engagement leads to enhanced nuclear factor kappa beta activation, upregulating RAGE expression in a positive feedback loop.30
 into plasma in healthy and Alzheimer")
Concomitantly, cytotoxic Aβ injures brain microvascular endothelial cells and critical tight junction-associated proteins, impairing BBB integrity.31 Recent studies have highlighted that decreased expression of claudins (CLDN-1 and CLDN-5) in AD patients augments the disruption of tight junctions and BBB with subsequent enhancement in Aβ accumulation following transporters dysfunction.31 At the same time, other studies have shown that approximately 50% of Aβ is transported across BBB using LRP-associated transcytosis and degradation of Aβ in smooth vascular cells.32 Soluble-LRPs (sLRP) in plasma are derived from the cleavage of LRP by beta-site amyloid precursor protein cleaving enzyme 1.33 Due to its high binding affinity with Aβ, sLRP is an essential endogenous component promoting the peripheral sink of Aβ.34 Notably, in the plasma of AD patients, sLRP is significantly oxidized, and shows decreased binding with Aβ.35
On the contrary, other evidence has shown a higher affinity of LRP-1 toward Aβ (1–42) and its upregulation in AD in a cell-dependent manner within neurons and astroglial cells containing senile plaques.36 P-glycoprotein (P-gp), a member of the ATP-binding cassette (ABC) transporters superfamily, is also expressed in the luminal surface of BBB endothelium and promotes the efflux of central nervous system active drugs.37 Interestingly, Aβ is a substrate of P-gp, and recent evidence has reflected its potential role in Aβ clearance.38,39 Decreased P-gp expression in AD and aging may impair Aβ clearance.40 During early AD, a compensatory rise in P-gp expression occurs to reduce Aβ accumulation; as the disease progresses, increased Aβ accumulation disrupts p-gP expression.41 In addition to these transporters, neonatal Fc receptors (FcRn) on BBB are associated with Aβ-transportation.42 FcRn also interacts with HSA and specific variants of immunoglobulins and regulates important FcRn-mediated recycling and transcytosis.43 Other members of the ABC family, including ABCA1, ABCG4, and ABCG2 (BCRP), have been found to transport Aβ from the brain to the peripheral blood.42
- 1.2
Transport of Aβ across blood-CSF barrier (BCSFB) – an insight into ‘CSF sink.’
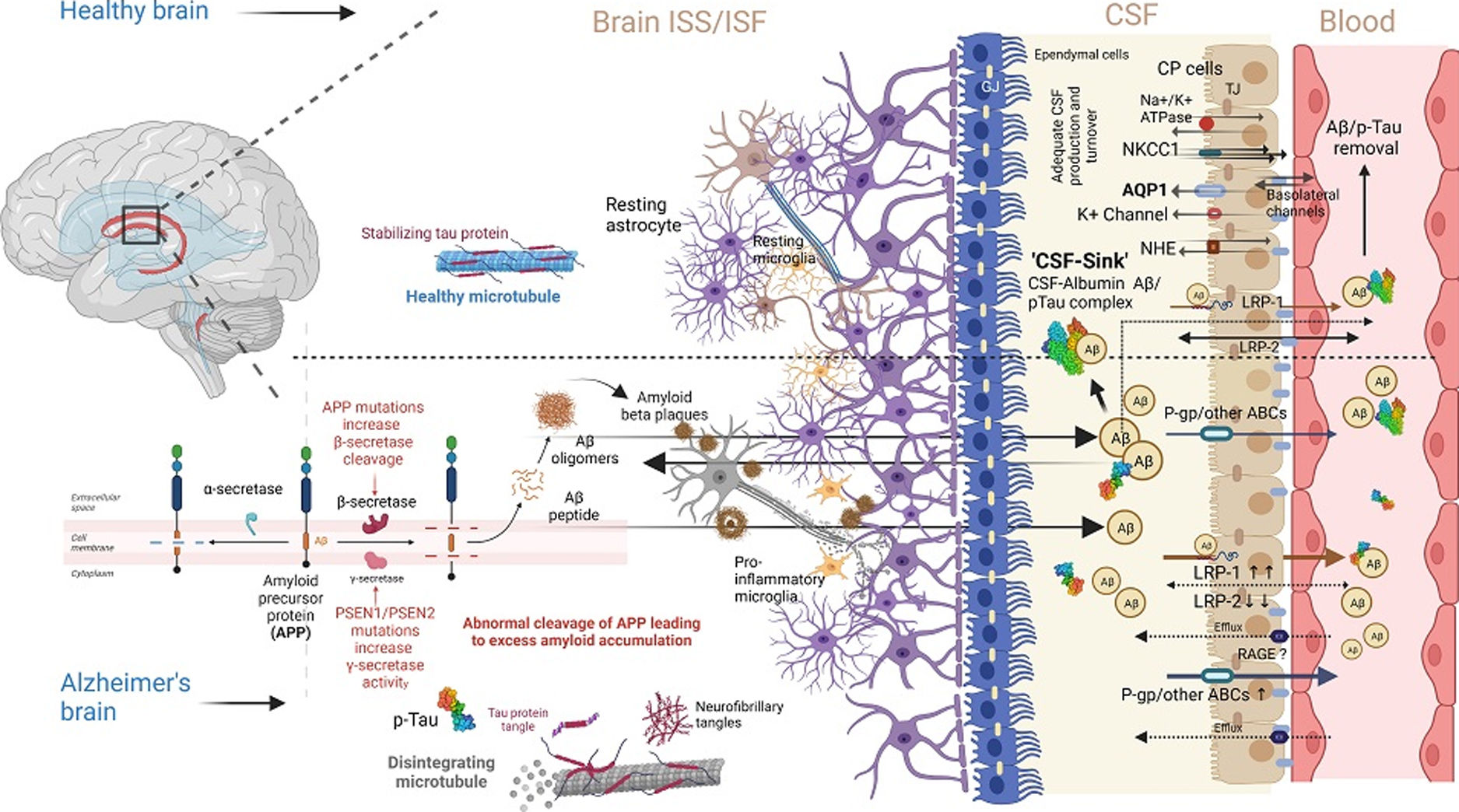
Aβ transportation across BCSFB is yet another essential avenue and a critical complementary half of Aβ – ‘peripheral sink,’ responsible for the tightly regulated three-point homeostasis of Aβ-load between interstitial brain space, plasma, and CSF23 (Fig. 2). Central to the function of BCSFB is the choroid plexus (CP).44,45 However, the contribution of CP in net Aβ-clearance is not substantial compared to BBB, which in turn emphasizes the importance of enzymatic Aβ degradation.46 CP is associated with neurogenesis in the injured brain.47 In AD, there is a deficit in CSF production and turnover rate, facilitating the excess accumulation of central nervous system toxic metabolites, including Aβ, along with the flattening of CP epithelium, basement membrane, and overall atrophy of CP epithelial cells.48–52 Importantly, Aβ transportation across BCSFB is bidirectional, and homeostasis decides the net flux of Aβ between Aβ-efflux and influx transporters.53 LRP-1 is also expressed in BCSFB epithelium along with BBB, as discussed. Like brain microvascular endothelial cells, LRP-1 targets Aβ for cellular enzymatic degradation and facilitates transcytosis across BSFB.54 sLRP-1 is also found in ISF, CSF, and plasma.55
 barrier (BCSFB) from brain interstitial space or fluid (ISS/ISF) into CSF in healthy and Alzheimer")
Interestingly, BBB-LRP1 expression is decreased in aging and AD, while BCSFB-LRP1 remains relatively more functional in active Aβ transportation out of the CSF.56,57 LRP-2, another member of the low-density lipoprotein receptor family, is a crucial Aβ transporter in CP epithelium, and its expression in CP is 17 folds higher than that of capillary endothelium.58 LRP-2 binds to apolipoprotein J and transports to CP epithelium, and its expression is significantly reduced at BCSFB with aging and AD.59 On this note, P-gp, a major efflux transporter in BBB, is also expressed in CP epithelium, but its role in Aβ transportation across BCSFB is unclear, just like the functional role of RAGE-like receptors in BCSFB in aging and AD.60,61 Besides, aquaporin 1 (AQP-1) expressed on the apical CP epithelial cells is significantly decreased in AD patients with a gradually decreasing CSF turnover rate as the disease advances.62
The glymphatic system comprises a network of extravascular channels that permits the circulation of CSF and interstitial fluid within and through brain parenchyma.63 It is a successful clearance machinery in removing extracellular Aβ.64 Recent studies have revealed that the glymphatic system can be a potential facilitator behind the clearance of senescent cells from the brain, making a connecting link between the glymphatic system and the extracranial lymphatic drainage pathway. In this regard, Li et al.65 demonstrated that the vascular endothelial growth factor C (VEGF-C)/C-C motif plays a crucial role in clearing senescent astrocytes via meningeal lymphatics, potentially contributing to AD pathogenesis. Neuroinflammation in AD results in impaired glymphatic clearance.66 The binding of soluble Aβ oligomer and Aβ with microglia receptors increases proinflammatory cytokines, eventually slowing the glymphatic flow.64,67 In addition to Aβ, the accumulation of tau tangles also negatively correlates with the coherence of the glymphatic system.64 Aquaporin-4 plays a significant role in brain fluid homeostasis, and the knockout model also showed decreased glymphatic flow.68 In this regard, the scientific community has opened the possibility that inhibitors of aquaporin-4 could be considered as a therapeutic approach to restore the glymphatic flow.
- 1.3
Clearance equilibrium of albumin-Aβ complex in serum and CSF: therapeutic plasma exchange in the restoration of ‘peripheral sink’ homeostasis.
Albumin is a crucial facilitator in maintaining Aβ ‘peripheral sink’ in conjugation with BBB, BCSFB Aβ transporters, and other plasma and CSF soluble-Aβ transporters or carriers.69,70 Aβ has a mean concentration of 0.1–0.5nM in plasma and CSF.71,72 Its systemic clearance depends on carrier proteins, including β2-macroglobulin, Apoliprotein-E, Apoliprotein-J (Clusterin), transthyretin, sLRP-1, and albumin.34,73–75 Unlike plasma or lymphatics, the CSF contains minimal albumin (∼0.025g/100ml), primarily through diffusion.76 Several studies have concluded that albumin, the most abundant protein in plasma and CSF, is the primary carrier of Aβ.77 However, several other pieces of evidence suggest that sLRP, the soluble cleaved protein of LRP1, could be a more efficient plasma Aβ carrier than plasma albumin.34 These contradictory statements and findings must be analyzed from the perspective of their binding kinetics. While some studies have supported 1:1 stoichiometric binding of monomeric Aβ with albumin, other findings suggest that albumin preferentially binds with oligomeric Aβ at its three different binding domains with a minimum dissociation constant (Kd).78–81 Hence, with a minimum Kd and 1:1 monomeric binding, albumin surpasses these contradictory findings, establishing a higher albumin affinity toward Aβ in plasma and CSF.82 Moreover, the sLRP-mediated peripheral sink is disrupted very early in patients with mild cognitive impairment (MCI) or MCI progressing to AD (MCI-AD) due to sLRP oxidative damage-mediated decreased plasma Aβ binding.35,83 Accordingly, chronically low serum albumin level is associated with poor cognitive function, which can further be aggravated in ApoE4 carriers.84 Besides, the inverse relationship between serum albumin and Aβ deposition is also evident from pre-clinical and clinical studies, which emphasizes the ‘Aβ shift’ from the brain into the plasma, driven by serum albumin.85,86 In this process, serum albumin binds with most of the Aβ in plasma and lowers the total Aβ load in the blood, balancing the dynamic equilibrium of Aβ between the brain and plasma.79,87 Supportive findings in favor of this tightly regulated dynamic equilibrium also emphasize a significant relation of low serum albumin with decreased Aβ binding capacity and increased plasma Aβ-load resulting in the inhibition of Aβ shift and increased brain parenchymal Aβ deposition.69,85 Long-standing low albumin also correlates with increased cerebrovascular risk.88 Moreover, Aβ bound to plasma albumin originates from several organs other than the brain, and these factors could alter the Aβ efflux and dynamic equilibrium between the brain and plasma.17
Notably, with progressing impairment of BBB-Aβ transportation in AD, BCSFB-Aβ exchange may prolong the Aβ-sink with an elimination rate of approximately 25%.89 This relatively less obstruction in BCSFB-Aβ transportation compared to plasma exchange is possible as there is no tight junctional barrier between CSF and brain parenchyma.90–93 Hence, factors in CSF have a wide range of effects on central nervous system. Interestingly, Aβ-transportation across BBB has an essential component of enzymatic degradation. Deposition of Aβ-plaques within brain-interstitial brain space displays similar first-order kinetics for isotope-labeled Aβ, in contrast to Aβ-transference from ISF to CSF due to the process of elimination, which is governed by CSF-turnover kinetics.94 Albumin is the most abundant protein in CSF, produced mainly through ultrafiltration of plasma protein and minimally by microglia.95,96
Of note is that the C-terminus domain of human-albumin shares a common sequence with apolipoprotein J or clusterin, an important Aβ-antiaggregatory factor in the brain.97 Studies have revealed that this domain at physiological CSF concentration is neuroprotective against Aβ (1–42) neurotoxicity, making it a critical albumin region.97 To add up, experimental findings from Ezra et al.98 reflected on the efficacy of adding albumin to CSF as a more direct way to eliminate toxic forms of brain parenchymal toxic Aβ intermediates, suggesting the emerging importance of CSF exchange with HSA replacement.
- 1.4
Diurnal patterns in neuropathologically hallmark molecules of Alzheimer's disease in CSF and serum: Implications of circadian influences on ‘transportation homeostasis.’
Sleep is considered a restorer for memory and cognition, especially the slow–wave sleep cycle, which has been found to consolidate long-term memory.99 Sleep disturbances in AD patients have been widely reported and found to be multifaceted, with incoherence in total sleep time, sudden nocturnal arousals, and reversal in the diurnal circadian pattern.100 Hence, it is imperative to understand the impact of sleep and its abnormalities toward Aβ-transportation homeostasis across BBB and BCSFB into maintained ‘peripheral sink’. It is also essential to understand how sleep restoration in AD patients with sleep abnormalities can re-establish homeostasis of Aβ load between brain parenchyma, plasm, and CSF.
Nir et al.101 hypothesized that diurnal variation in CSF-Aβ load is possibly associated with neuronal hyperactivity during wakefulness, whereas decreased neuronal activity was observed during slow-wave sleep. Huang et al.102 revealed higher levels of CSF Aβ (1–40) and Aβ (1–42) during wakefulness in amyloid positron emission tomography positive non-AD in comparison to amyloid positron emission tomography negative non-AD patients of more than 60 years. Several pieces of evidence have pointed toward Aβ-42 sequestration into plaques of older individuals with positive amyloid deposition, eventually resulting in decreased clearance of Aβ into ISS, less diurnal Aβ-concentration variation, and altered neuronal firing.103 Besides, Aβ-oligomers and tau production may be relatively accelerated due to loss of slow-wave sleep secondary to aging or sleep abnormalities and sleep-breathing disorders, eventually leading to sleep-phase alteration with increased wakefulness during sleep.104 Hence, we can hypothesize that improving sleep quality by increasing the slow-wave sleep phase by treating underlying sleep disorders with medications and behavioral interventions can effectively reduce Aβ levels and its associated long-term risk of developing AD. So, why does Aβ accumulation prolong wakefulness and alters regular sleep pattern? Moreover, a deeper understanding is needed to understand how the dynamic relation between sleep-wake patterns and cognitive status maintains transportation homeostasis across blood and CSF barriers.
Furthermore, the diurnal CSF-Aβ variation pattern is consistent with hypocretin's diurnal fluctuation, a wakefulness regulator.105 A study by Kang et al.105 has also revealed that orexin displays a circadian rhythm in both AD and healthy individuals.106 Besides, CSF-Aβ concentration increases during orexin infusion and decreases with orexin-receptor antagonist, pointing toward a potential role of orexin and sleep-wake cycle dissociation in the pathogenesis and progression of AD.107
Independent of the sleep–wake cycle, diurnal variation of AD biomarkers in CSF is a matter of considerable interest and debate as it has diagnostic and therapeutic implications in neurodegenerative diseases. It is already known that almost 1/3rd of individuals presenting with AD may show negative Aβ.108 In the current era of clinical trials, amyloid status is an essential inclusion criterion, notably for anti-amyloid antibodies. However, it is still unknown if this negative biomarker status relates to the diurnal variation of Aβ in the body fluids. Such investigations are technically challenging as this might require admitting individuals to the in-patient department and collecting serial CSF samples by achieving recurrent lumbar punctures on a single day. A recent study has recently addressed this question by performing serial measurements of CSF amyloid beta on 13 patients. Although a significant variation was found among different individuals, no variation of CSF Aβ was reported at different time points on the same day, indicating possible diurnal stability.109
- 2.
Molecular interactions between albumin and neuropathologically hallmark molecules of Alzheimer's disease:
Following our previous sections, the effect of HSA with various forms of Aβ, including Aβ (1–40) and Aβ (1–42), has been widely reported and studied. While different subtypes of Aβ-isoforms are present in the brain parenchyma, the hydrophobicity of Aβ (1–42) is particularly prone to self-aggregation into soluble oligomers, which may further form larger fibrils and accumulate into insoluble extracellular plaques with increasing neurotoxicity.110,111 Precisely, robust evidence on the toxic potential of soluble Aβ-oligomers showed that it inhibits electrophysiological and ultrastructural mechanisms of synaptic plasticity by reducing long-term hippocampal potentiation and dendritic spine density.112 Besides, it also causes a rapid decrease in the membrane expression of memory-related receptors, along with abnormal spine morphology and synaptic deterioration in cultures of hippocampal neurons.113 Experimental data have also suggested that the biophysical nature of Aβ-oligomers with low molecular weight and high aqueous diffusibility further potentiates the destruction of neurite integrity and functionality, even in a small quantity.114 Importantly, due to the dynamic nature of Aβ-oligomers and the lack of specific molecular tools for each assembly, it has been challenging to analyze Aβ-oligomers in human brain tissue and in the brains of AD mouse models.115 Despite this challenging obstacle, several studies have come up with reliable identification of several specific Aβ-oligomers species by combining immunological and biochemical principles.116
Kuo et al.79 and others117 experimented on the affinity of monomeric Aβ with HSA, which resulted in the determination of dissociation constant (Kd) in the range of 5–10μM for both Aβ (1–40) and Aβ (1–42), based upon a 1:1 stoichiometric mixing ratio. Similarly, in the case of the Aβ-42 peptide, frequent monomeric interactions between their Q15-V24 and N27-V36 regions resulted in the formation of β-hairpin, which is critical for β-sheet association and aggregation. Computational-based studies have simulated the interactions between Aβ42 and HSA regions using replica exchange with solute tempering. They have reported binding of Aβ42 on multiple sites of HSA with a preference for domain III and changes in various conformations of Aβ42 from their free state. These bindings significantly reduce the β-sheet abundances of the H14-E22 and A30-M33 regions and β-hairpin rich regions. The formation of promiscuous interactions at the edge of the peptide-protein interface between HSA and the hydrophobic cores of Aβ42 at the center disrupts intrapeptide interactions crucial for β-sheet formation.118
Further, into the interactions of Aβ-oligomers and fibers, studies using Aβ oligomer-albumin complexes with off-resonance relaxation magnetic resonance experiments tagged with dynamic light scattering followed by domain deletions and ultrafiltration were done. They have shown that Aβ-oligomers were recognized by HSA through evenly partitioned sites, which are similar across three albumin domains that bind to Aβ-oligomers with similar dissociation constants in the nanomolar range based on a Scatchard-like model of the albumin inhibition isotherms. These data also convey that a single high-affinity albumin-binding site per Aβ protofibril prevents the further extension of insoluble Aβ aggregates in the presence of HSA.81
Focusing on the sub-structural interaction of peptides (Aβ) and proteins (HSA), the inhibitory effects of HAS on Aβ-oligomers are evident; HSA (residue: 494–515) recognizes the central hydrophobic core of Aβ and results in inhibition of Aβ self-assembly.119 Finally, to summarize the interactions and verify their effects in vivo, studies using molecular docking and SHSY5Y neuroblastoma cells have shown that these interactions can inhibit Aβ aggregation and promote the disassembly of Aβ aggregates, thereby creating a neuroprotective effect. They found that the C-terminus region of HSA was responsible for the impairment of Aβ aggregation, as demonstrated through reverse-phase chromatography and electron microscopy analysis at micromolar concentrations in vitro. MTT-based cell viability assays also demonstrated the neuroprotective role of C-terminus albumin against Aβ 1-42 oligomers and fibers.97 Therefore, we can imply that these interactions between HSA and Aβ are significantly meaningful pathophysiologically and can be implied as therapeutic targets for dementia patients. Their interactions are well-studied in vitro, but practical applications and in vivo techniques are required to validate the in vitro data further.
Tau protein is critical in stabilizing, assembling, and maintaining microtubules and optimum neuronal functions.120 During diseased conditions, many pathological modifications of the tau protein occur.121 These changes lead to tau protein aggregation and the formation of paired helical filaments and neurofibrillary tangles (consisting of hyperphosphorylated tau), the hallmarks of AD and other tauopathies.122,123 Evidence has established that tau mediates the neurotoxic effect of Aβ-oligomers, and the pathological changes in tau biology are associated with sub-cellular localization, which might be responsible for neuro-cellular derangement and cognitive decline.124 The interactions of Aβ plaques and neurofibrillary tangles are well studied for their synergistic interactions, which lead to neural damage and enhanced AD pathology.125,126 Aβ and tau cross-seeding are extensively studied under in vitro conditions and their pronounced cytotoxic effect on neuronal cell damage.127,128 Interactions between Aβ and HSA have also shown positive findings, though their clinicopathological implications are not yet fully understood. Several studies have shed light on HSA-driven Aβ clearance and cross-seeded amyloid-tau plaque clearance under regulated experimental conditions.129,130
Notably, chronic exposure to Aβ in experimental models’ brain tissue leads to transactive response deoxyribonucleic acid binding protein of 43 kDa (TDP-43) pathology, and TDP-43 can further augment the seeding of Aβ into oligomeric Aβ.131 However, some evidence has revealed that TDP-43 inhibits Aβ-fibrilization at the initial and oligomeric stages but not in the fibrillar stage.124 In experimental models, it enhances Aβ’s ability to cause long-term hippocampal potentiation impairment upon intrahippocampal injection, resulting in spatial memory defect.132 These experimental findings may correlate with more severe memory loss and extensive hippocampal atrophy in AD patients with TDP-43 inclusions.133,134 Shih et al.135 showed that TDP-43 oligomers could be co-localized largely with intraneuronal Aβ and some with amyloid plaques in APP/PS1ΔE9 mice and AD brains. Besides, it is also essential to understand the intracellular interaction sites between TDP-43 and Aβ, which could be in the cytoplasm or lysosome and have been reported to alter neuro-cellular endosomal-lysosomal pathways, triggering the autophagy mechanism due to their aggregation.136,137
Though the relationship between Aβ and TDP-43 lacks profound understanding, it is known that TDP-43 co-pathology is much more common in AD neuropathogenic changes than in tauopathies, such as frontotemporal lobar degeneration-tau, progressive supranuclear palsy or cortico-basal syndrome138; the interaction between Aβ and TDP-43, is imperative but needs more profound understanding. Additionally, evidence on the relationship between TDP-43 and tau-pathology in AD patients is intriguing. Joseph et al.139,140 described higher Braak staging of tau pathology in patients with TDP-43 co-morbidity. Hence, further understanding of the synergism among Aβ, tau, and TDP-43 and its impact on these molecules’ transportation homeostasis and dynamics can elucidate new therapeutic strategies in AD.
However, the interactions of tau and its phosphorylated residues and TDP-43 with HSA are poorly understood and need further experimental explanation. Nonetheless, we can hypothesize that HSA can clear cross-seeded amyloid plaques due to its robust interactions with Aβ and provide therapeutic benefits in AD patients.
- 3.
Immunomodulatory effects of HSA:
HSA has been found to positively modulate neuro-protection, neuronal and neuro-glial coupling, and barrier maintenance. Its immunomodulatory function in various subdivisions of the immune system comprises innate and adaptive immunity. As far as the innate immune system is concerned, it has been found to regulate the inflammatory mechanism and leukocyte activity.141,142 Subsequently, it strongly associates with the expression of major histocompatibility complex-II (MHC-II) molecules. An in vitro antigen presentation assay with murine P3888D1 cells and bone marrow-derived dendritic cells manifested elevated expression of major histocompatibility complex-II molecules, eventually leading to helper T-cell activation and subsequent interleukin-2 secretion due to therapeutic preparation of HSA.141 The study mentioned above also states that the regulatory effect of HSA is dose-dependent. Several studies have found the interplay between albumin and C-reactive protein (CRP) levels to be significant.143 A study by Sheinenzon et al.142 of 4344 patients reported a negative correlation between CRP and albumin, i.e., relatively higher CRP in the case of hypoalbuminemia.
Human serum albumin contains at least seven fatty acid binding sites, with three high-affinity binding sites firmly binding to glycerol monolaurate. This fatty acid monoester shows binding solid affinity and low micromolar dissociation (Kd∼1.4μM) under isothermal titration calorimetry.144 The mechanism of immunomodulation by HSA lies in the activation of the AKT signaling pathway. Activation of AKT requires upstream activation of PI3K and, subsequently, phosphorylation of AKT at both threonine 308 and serine 473 sites, which glycerol monolaurate hinders, leading to its immunosuppressive property.144,145 The oligomeric-Aβ is believed to generate toxicity by enhancing calcium ion influx into the neuronal and neuroglial cells, leading to cellular stress and subsequent degeneration.146,147 Of note is that SHSY5Y cells treated with HSA show a reduced influx of calcium ions compared to the cells without HSA upon exposure to the calcium-sensitive dye Fluo-AM, followed by o-Aβ.97
It is noteworthy to mention microglial macrophage-inducible C-type lectin (Micle), a specific innate immune factor activated to sense necroptotic cell death upon binding with a subunit of histone deacetylase named Sin3A associated protein 130 during subarachnoid hemorrhage. The prognosis of subarachnoid hemorrhage is negatively related to the extent of post-hemorrhagic inflammatory response and neutrophil influx, which is significantly influenced by Micle/syk (spleen tyrosine kinase) downstream pathway.148 A study by Xie et al.148 showed that albumin promotes its anti-inflammatory response upon binding with the Micle receptor and deactivates the downstream CARD9/Bcl-10 and NLRP3/Caspase-1 pathways followed by the inhibition of Sin3A associated protein 130.
- 4.
Clinical and pre-clinical evidence of plasma exchange and albumin replacement in dementia and neurodegeneration:
Several clinical and pre-clinical studies and trials over the years have been conducted through different approaches to the possible efficacies of therapeutic plasma exchange and albumin replacement in patients with Alzheimer's and non-Alzheimer types of dementia. The current trend in therapeutic targets for AD has been approached with a particular focus on anti-amyloid and anti-tau-based treatment development. So far, limited clinical benefits have been observed in these randomized controlled trials with therapies targeted toward amyloid cascade.149 In this regard, immuno-therapeutics targeting Aβ oligomers (aducanumab) have displayed encouraging results, yet most of the others have not proven apparent efficacy.150 Importantly, monomeric, oligomeric, proto-fibrillary, and fibrillary forms of Aβ are in equilibrium, and targeting any component augments the formation of the others.151 A recent addition to the monoclonal-based therapy includes lecanemab, which shows relatively high selectivity toward soluble aggregates of Aβ compared to monomeric amyloid. It has been associated with mild to moderate incidences of amyloid-related imaging abnormalities, including edema or effusion and cerebral hemorrhage.152 Altogether, it points out the importance of testing new therapeutic avenues.
On this avenue, the Alzheimer management by albumin replacement (“AMBAR”) study was initiated with a rationale of lowering soluble Aβ by augmenting and restoring ‘peripheral-sink’ hypotheses as discussed above, as well as by removing plasma and its replacement with therapeutic grade human albumin and intravenous immunoglobulin.153 AMBAR is a multicentric, randomized, double-blinded, placebo-controlled study in which all recruited patients (n=496) with mild to moderate AD were treated for 14 months. All treated patients initially underwent removal of 2500–3000ml of plasma (high plasma exchange group), with an equivalent replaced volume of 5% human serum albumin weekly for six weeks through peripheral or central venous route followed by monthly plasma exchange for 12 months.153 Within these included patients, 690–880ml of plasma (low volume plasma exchange group) was removed and replaced with 100ml of 20% HSA (20g) plus 200ml of intravenous immunoglobulin (low plasma/low intravenous immunoglobulin group) or 200ml of 20% [40g] plus 400ml of intravenous immunoglobulin (high albumin/high intravenous immunoglobulin group).153 The second phase of the plasmapheresis was performed only via the peripheral route.153 The study's outcome reflected that the treatment group was associated with an average of 47–70% less worsening in AD assessment scale-cognitive subscale (ADAS-Cog) scores and 42–75% less worsening in AD Cooperative Study-Activities of Daily Living (ADCS-ADL) score than the control subjects.153 Notably, along with the deceleration of the disease progression in the treated patients with mild AD, a pretty unexpected improvement was found in placebo-treated mild-AD patients, making the therapeutic efficacy statistically inconclusive.153 AMBAR's findings raise the most critical question: what possible mechanisms could slow the disease progression?,153 Amongst the plausible theories, reduction in neurotoxic Aβ species, tau-pathology, reduced neuroinflammation, oxidative stress, and microcirculatory deficit are significant and have been discussed in previous segments. Neuroimaging analyses in mild to moderate AD patients have been performed in the recent AMBAR phase 2b/3 trial with MRI volumetric, regional, and statistical parametric mapping analysis on positron emission tomography with 18F-fluorodeoxyglucose.154 Plasma exchange with albumin replacement was associated with much lower debilitating changes in subcortical structures and less metabolic decline than in AD. This finding was even more marked in moderate-AD patients treated with high albumin and intravenous immunoglobulin compared to low albumin with or without intravenous immunoglobulin.154
Plasma is rich in diverse antibody spectrum and other least understood beneficial factors, which may be an essential source to search and develop therapeutics against AD. Sha et al.155 reported the efficacy of young fresh frozen plasma infusion in mild-moderate AD with better performance in ADCS-ADL scores than placebo. As discussed, it is essential to note that the plasma level of β-2-microglobulin and other proinflammatory factors, including CCL-11, could be decreased by therapeutic plasma exchange, providing beneficial effects in AD.156 Young plasma infusion has been efficacious in old mice models by improving learning and memory function.156 Interestingly, naturally occurring autoantibodies against Aβ, which comprise approximately two-thirds of the total human antibody pool, are significantly reduced in patients with AD. Natural autoantibodies against Aβ are associated with the inhibition of Aβ fibrilization, reduction in toxicity, and increased Aβ clearance.157,158 However, this naturally occurring antibody pool's underlying principles are not understood. Utilizing this idea, Dodel et al.159 showed the possible efficacy of intravenous immunoglobulin treatment in mild to moderate AD patients. Simultaneously, Kile et al.,160 in their randomized double-blinded 24-month-long study, reflected the clinical efficacy of intravenous immunoglobulin treatment in the incidence of brain atrophy and the conversion of mild cognitive impairment to AD. Pre-clinical studies on plasma fractions have revealed good efficacy on brain aging with fresh frozen plasma and showed superior and long-term modulatory effects on neurogenesis.161 A study of GRF6019 in 47 mild-to-moderate AD patients showed improved cognitive scores in the 250ml GRF6019 group.162 A phase-2 study of GRF6021 conducted on 89 patients with Parkinson's disease dementia for six months showed improvement in executive function, verbal abstraction, and orientation.163 Kim et al.164 collected plasma from young exercised mice and infused 100μ-liter of this plasma into 12 months AD (3×Tg-AD) 10 times at 3-day intervals and compared it with plasma from young mice that did not exercise. Mice infused with plasma from young exercised mice had higher plasma brain-derived neurotrophic factor levels, improved memory and mitochondrial function, less cell death in the hippocampus, and a higher level of synaptic proteins. Khrimian et al.165 reported that infusion of 1-month-old mice with 3-month-old plasma eight times over 24 days improved memory on a novel object recognition task and reduced anxiety on an elevated plus maze task. However, there were no memory improvements when old mice were infused with plasma from osteocalcin knockout (Ocn −/−) mice. Memory was impaired when young mice were treated with an osteocalcin-neutralizing antibody, and memory improved when these old mice were treated with osteocalcin over two months.165 Tang et al.166 treated middle-aged with young and undamaged recombinant serum albumin (1.5mg/g) every three weeks. This recombinant therapy improved learning and memory on the Barnes maze test in male mice and reduced levels of p-tau in male mice but not females.
Mehdipour et al.167 conducted a study on ‘neutral’ blood exchange with saline containing 5% albumin after replacing plasma from animals and observed that old mice improved hippocampal neurogenesis compared to older mice that had undergone heterochronic blood exchange. Katsimpardi et al.168 showed that heterochronic parabiosis experiments between 2-month-old wild-type and 15-month-old mice increased neurogenesis in older ones without any effect in younger mice. Ghosh et al.169 reported that parabiosis between old and young mice over four weeks reduced inflammatory markers (monocyte chemoattractant protein-1, interleukin-6 and CRP) and senescence markers (p16, p21) in adipose tissue of old mice. In vitro, administration of young plasma to aged adipose explants also reduced markers of senescence and some inflammatory markers, mostly tumor necrosis factor-α and monocyte chemoattractant protein-1 but not interleukin-6. Rebo et al.170 interestingly showed a heterochronic blood exchange paradigm in small animals that did not require connecting the circulatory system of the animals. The homogenization of blood between two animals showed no difference in neurogenesis in older animals, while the young animals that received old blood had a reduction in neurogenesis. This suggests that different tissues may respond differently to young and old factors.
A study by Wang et al.171 exploring the correlation between serum albumin levels and the risk of MCI showed that subjects with low serum albumin had more than a 100% increased risk of MCI compared to those with normal serum albumin levels. This study also explored the role of serum albumin on the possible etio-pathological risk factor of MCI in subjects with specific conditions, including arterial hypertension, metabolic syndrome, cardiovascular diseases, and cerebrovascular disorders and found positive synergism. Mizrahi et al.172 interestingly revealed a possible intercorrelation between serum albumin and cognitive dysfunction in elderly patients with hip fractures, with lower serum albumin associated with a lower mini-mental state examination score.
Min et al.173 found that chronically low serum albumin was associated with poor cognitive functions in Korean veterans and their families with low overall mini-mental state examination scores. Older adults with APOE-E4 alleles showed higher association than those without this allelic variance, reflecting the negative associations of APOE4 carriers with lowered albumin levels on cognitive function and brain aging. A national population-based survey study from the UK revealed that older populations with lower albumin levels exhibited poorer cognitive functions than younger individuals174. Ng et al.175 showed that lower serum albumin level was significantly associated with lower mini-mental state examination scores in older Chinese adults irrespective of age, sex, education, and vascular risk factors. Their subsequent follow-up study confirmed these cross-sectional findings with a more significant decline in cognitive performance in those elderly subjects with lower serum albumin. Notably, APOE is a primary cholesterol carrier in the brain and shows extensive polymorphic variations. Despite its established correlation as a risk factor for AD, detailed insight into the association of APOE4 with cognitive impairment is still a gray area.176 Ng et al.175 found a pronounced and intriguing association between APOE4 carriers and non-carriers with low average albumin in this context. However, the ‘Albumin-APOE-Cognitive decline’ mechanism is unclear and needs additional studies.
Wu et al.177 found dementia severity was significantly associated with kidney function, serum albumin, and hemoglobin in the oldest-old with AD. However, the restoration of renal function in these patients, along with the restoration of albumin and its possible correlation with cognitive improvement in the older population, requires further studies. On this note, Takae et al.,178 using data from the ‘Hisayama study’, revealed that albuminuria increases the risks for all-cause dementia, AD, and vascular dementia in Japanese elderly independent of other potential risk factors for an abnormal. A study by Wang et al.179 also revealed that individuals with lower estimated glomerular filtration rates are at higher risk of incident vascular dementia. A study in dementia patients with Aβ deposition revealed that they had significantly higher indirect bilirubin concentrations, lower HSA levels, and a corresponding higher indirect bilirubin/HSA ratio. It was also found that there was significant improvement in ADCS-ADL and the sum of boxes of the clinical dementia rating scores in patients with mild to moderate dementia following intravenous infusion of human albumin.180 Koyama et al.181 showed a significant correlation between malnutrition and cognitive disorders in older populations with lower serum albumin and hemoglobin among patients with diffuse Lewy body disease and frontotemporal dementia than the AD group in comparative age groups. It is noteworthy to mention that a recent pilot study by Povedano et al.182 showed the efficacy of plasma exchange with 5% albumin replacement in amyotrophic lateral sclerosis patients, with 63.4% displaying a slower-than-expected decline in their Amyotrophic Lateral Sclerosis Functional Rating Scale score. Taking together, a multidimensional nutrition assessment in these patients with dementia would need future exploratory studies.
Table 1 summarizes the interventional plasma exchange and albumin replacement trials in patients with AD and non-AD dementia and neurodegenerative conditions.
- 5.
Clinical perspective and future possibilities of therapeutic plasma exchange in dementia
Interventional plasma exchange and albumin replacement trials in patients with Alzheimer's and non-Alzheimer's dementia and neurodegenerative conditions.
| Author [reference] | Study title | Trial registry authority | Study design or type | Sample size/dementia rating | Study outcome | Grade certainty rating |
|---|---|---|---|---|---|---|
| Boada et al.153 | A randomized, controlled clinical trial of plasma exchange with albumin replacement for Alzheimer's disease: primary results of the AMBAR Study. | EudraCT#: 2011-001598-25; ClinicalTrials.gov ID: NCT01561053. | Phase 2b/3 trial examining the effects of plasma exchange in patients with mild-to-moderate Alzheimer's disease. Different doses of albumin and intravenous immunoglobulin replacement (6-week period of weekly conventional plasma exchange followed by a 12-month period of monthly low-volume plasma exchange), and placebo (sham). | Three hundred forty-seven Alzheimer's disease patients (496 screened) were randomized into three plasma exchange treatment arms. | Plasma exchange-treated patients performed significantly better than placebo for the co-primary endpoints: change from baseline of Alzheimer's Disease Cooperative Study-Activities of Daily Living (P=0.03; 52% less decline) with a trend for Alzheimer's Disease Assessment Scale–Cognitive Subscale (P=0.06; 66% less decline) scores at month 14. Moderate- Alzheimer's disease patients (baseline Mini-Mental State Examination 18–21) scored better on Alzheimer's Disease Cooperative Study-Activities of Daily Living (P=0.002) and Alzheimer's Disease Assessment Scale–Cognitive Subscale (P=0.05), with 61% less decline in both. There were no changes in mild Alzheimer's disease patients (Mini-Mental State Examination 22–26). Plasma exchange-treated patients scored better on the Clinical Dementia Rating Sum of Boxes (P=0.002; 71% less decline) and Alzheimer's Disease Cooperative Study-Clinical Global Impression of Change (P<0.0001; 100% less decline) scales. This trial suggests that plasma exchange with albumin replacement could slow cognitive and functional decline in Alzheimer's, although further studies are warranted. | Medium |
| Sha et al.155 | Safety, tolerability, and feasibility of young plasma infusion in the plasma for Alzheimer's symptom amelioration study: a randomized clinical trial. | ClinicalTrials.gov ID: NCT02256306. | The Plasma for Alzheimer Symptom Amelioration (PLASMA) study randomized nine patients under a double-blind crossover protocol to receive four once-weekly infusions of either one unit (approximately 250ml) of young fresh frozen plasma from male donors or 250ml of saline, followed by a 6-week washout and crossover to four once-weekly infusions of alternate treatment. After an open-label amendment, nine patients received four weekly young fresh frozen plasma infusions, and their subjective measurements were unmasked. | Eighteen consecutive patients with probable mild to moderate Alzheimer's disease, a Mini-Mental State Examination (score of 12–24 inclusive), and an age of 50–90 years were enrolled. Thirty-one patients were screened. Thirteen were excluded (11 failed the inclusion criteria, and two declined to participate). | There was no difference in the age (mean [SD], 74.17 [7.96] years), sex (12 women [67%]), or baseline Mini-Mental State Examination score (mean [SD], 19.39 [3.24]) between the crossover (n=9) and open-label groups (n=9). There were no related serious adverse events and no statistically significant difference between the plasma (17 [94.4%]) and placebo (9 [100.0%]) cohorts for other adverse events, which were mild to moderate in severity. The young fresh frozen plasma treatment was safe, well tolerated, and feasible. The study's limitations were the small sample size, short duration, and change in study design. | Low |
| Hannestad et al.162 | Safety and tolerability of GRF6019 in mild-to-moderate Alzheimer's disease dementia. | https://clinicaltrials.gov/study/NCT03520998. | Forty-seven patients were randomized to receive daily infusions of 100ml (n=24) or 250ml (n=23) of GRF6019 for five consecutive days over two dosing periods separated by a treatment-free interval of 3 months. | Enrollment eligibility required a diagnosis of probable Alzheimer's disease, age 60–90 years, and a Mini-Mental State Examination score of 12–24. | The mean (SD) baseline Mini-Mental State Examination score was 20.6 (3.7) in the 100ml group and 19.6 (3.7) in the 250ml group; at 24 weeks, the within-patient mean change from baseline was −1.0 points (95% confidence interval [CI], −3.1 to 1.1) in the 100ml group and +1.5 points (95% CI, −0.4 to 3.3) in the 250ml group. The within-patient mean change from baseline on the Alzheimer's Disease Assessment Scale-Cognitive subscale was −0.4 points (95% CI, −2.9 to 2.2) in the 100ml group, while in the 250ml group it was −0.9 points (95% CI, −3.0 to 1.2). The within-patient mean change from baseline on the Alzheimer's Disease Cooperative Study-Activities of Daily Living was −0.7 points in the 100ml group (95% CI, −4.3 to 3.0) and −1.3 points (95% CI, −3.4 to 0.7) in the 250ml group. GRF6019 was safe and well tolerated, and patients experienced no cognitive or minimal functional decline. | Low |
| Michael J. Fox Foundation for Parkinson's Research Study Director: Alkahest Medical Monitor, Alkahest, Inc.163 | A study to assess the safety of GRF6021 infusions in subjects with Parkinson's disease and cognitive impairment. | ClinicalTrials.gov ID: NCT03713957. | Subjects received 250mg of GRF6021 five consecutive days in weeks one and 13.Others received a placebo for five consecutive days at weeks one and 13. The study period was approximately 24 months. | Fifty-three Parkinson's disease patients with cognitive impairment received GRF6021, and 26 received a placebo. | No serious adverse events were attributed to GRF6021, and more than 80% of participants completed both infusion regimens. Headache and falls were more common in the GRF6021 group compared to placebo. The trial reported improvements in some secondary endpoints. The Montreal Cognitive Assessment and the PDQ-39 quality of life measure showed small but statistically significant improvements from baseline, while the placebo group did not change. | Low |
| Zhong et al.180 | Abnormal serum bilirubin/albumin concentrations in dementia patients with Aβ deposition and the benefit of intravenous albumin infusion for Alzheimer's disease treatment. | The Affiliated Brain Hospital Ethics Committee of Guangzhou Medical University (Guangzhou Huiai Hospital) approved this study. The intervention study was registered at http://www.chictr.org.cn (ChiCTR-IOR-17011539). | Bilirubin and albumin concentrations in dementia patients with Aβ deposition were examined. Cell viability and apoptosis were determined in dopaminergic neuron-like cells MN9D treated with bilirubin in diverse serum concentrations. Human albumin at a dose of 10g every two weeks for 24 weeks was administered intravenously to Alzheimer's disease patients to examine the effect of albumin on Alzheimer's disease symptoms. | Eligible participants, who were between 50 and 90, met the standardized criteria for probable mild or moderate Alzheimer's disease, scored 10 and 26 on the Mini-Mental State Examination, and had normal liver function. | Significantly higher indirect bilirubin concentrations, lower albumin concentrations, and a higher ratio of indirect bilirubin to albumin were observed in dementia patients with Aβ deposition, including Alzheimer's disease, dementia with Lewy bodies, and general paresis of insane. Analysis of the combined data of the entire 28 weeks of the assessment period using the area under curve showed significant improvements in the change of albumin concentrations, Alzheimer's Disease Cooperative Study-Activities of Daily Living scores, and Clinical Dementia Rating Sum of Boxes scores. Indirect bilirubin and indirect bilirubin to albumin ratio was significantly higher in dementia patients with Aβ deposition; intravenous albumin administration may benefit Alzheimer's disease. | Low |
| Povedano et al.182 | Plasma exchange with albumin replacement and disease progression in amyotrophic lateral sclerosis: a pilot study | ClinicalTrials.gov ID: NCT02479802Eudract number: 2013-004842-40 | Primary endpoints were changes from baseline in the Amyotrophic Lateral Sclerosis Functional Rating Scale-Revised score and forced vital capacity through 48 weeks. A post hoc analysis compared individual patient data with the expected Amyotrophic Lateral Sclerosis Functional Rating Scale-Revised progression slope. | Open-label, non-controlled, single-arm, prospective pilot study.13 adults with amyotrophic lateral sclerosis had six months’ treatment with plasma exchange with 5% albumin replacement and a six-month follow-up | The median Amyotrophic Lateral Sclerosis Functional Rating Scale-Revised score declined throughout the study, although the rate of decline was slower than expected in seven patients at the treatment end and five patients at the study end. Six patients remained in the same baseline slope progression category; four improved their slope category at the treatment end. Median forced vital capacity decreased significantly during the study. Treatment was well tolerated. Of 330 plasma exchanges with 5% albumin replacement procedures, 0.9% were associated with potentially related adverse events. Although functional impairment progressed, about two-thirds of patients showed a slower-than-expected rate of decline at the treatment end. Most patients had unaltered (54.5%) or reduced (36.4%) Amyotrophic Lateral Sclerosis Functional Rating Scale-Revised slope progression at treatment end. Further evaluation of plasma exchange with 5% albumin replacement in controlled studies involving more patients is warranted. | Low |
Plasma replacement studies in experimental mouse models of AD suggest a wide range of unknown factors in the blood that can worsen, improve, and attenuate neurodegenerative pathology and cognitive performance. However, these findings need to be translated into therapeutic applications and treatment strategies. Clinical results obtained from therapeutic plasma exchange trials in MCI-AD patients and observational cross-sectional studies on underlying chronic disease-associated dementia have several methodological flaws that can potentially undermine important correlating parameters and their applicability in AD groups. The limited patient size of the available studies may have implications on the homogeneity of treatment group at baseline favoring specific treatment arms, as clearly evident in the AMBAR study design with a more consistent cohort of patients but with a complicated study design involving four treatment groups and two study phases.183 Besides, these initial studies, including the AMBAR trial, did not incorporate biomarker-proven (amyloid positron emission tomography scan/CSF-Aβ-levels) AD cases at the entry-level.184 Notably, in the AMBAR study, 28% of recruited patients did not show evidence of Aβ brain deposition, which reflects that many included patients had other types of dementia. Simultaneously, it is equally important to consider more appropriate ways to protect the study blind group and the appropriateness of the study control group. Besides, the exact role of therapeutic plasma exchange, apprehended through most of these clinical trials as augmentation the cerebral Aβ clearance in early dementia, needs firm establishment in clinical settings.185 In previous segments, we have mainly discussed the efficacy of therapeutic plasma exchange by considering different activated plasma components, including HSA in vitro and experimental studies. However, the true nature of such interaction in clinical models needs further studies. On this note, a growing body of researchers has investigated the effect of this ‘young blood’ on the aging brain, suggesting that a more youthful systemic milieu might alter the course of brain aging and related dysfunction.186 Therapeutic plasma exchange could be helpful in the removal of inflammatory mediators, antibodies, reactive oxygen species, and related senescence factors from the plasma of AD patients.
Factors present in the young plasma, as discussed through some critical pre-clinical studies, may have beneficial properties that can modulate the neuroprotective mechanism by restoring barrier dysfunction, neurovascular conjugation, and the exchange process. It could also be that therapeutic plasma exchange with albumin replacement positively augments the removal of several detrimental factors from within the plasma of AD patients, such as β2-microglobulin or other pro and neuro-inflammatory chemokine/cytokines. Supplemental benefits of albumin and intravenous immunoglobulin could have elemental beneficial effects by suppressing these detrimental triggers. However, therapeutic plasma exchange in AD patients, especially in older people, may be associated with related complications, including acute lung injury, circulatory overload, anaphylactic reactions, and transfusion-related reactions.8,187 Nevertheless, regular clinical practice sufficiently manages most of these expected complications.
ConclusionsThe clinical evidence supporting therapeutic plasma exchange in dementia is minimal and needs further development to outweigh adverse effects and impact on health-related quality of life. Applying cutting-edge molecular research and clinical interventions has yielded insight into a broad spectrum of cellular and molecular targets of origin of pro-aging and pro-youthful factors in serum. Several of these factors promise to reduce the risk of dementia and related neurodegenerative diseases, including albumin as the most abundant protein in plasma. It is unmistakable that the combination of plasma removal through plasmapheresis and replacement with HSA has produced a significant encouraging result. It is essential to conduct extensive and fully masked research studies with bio-marker confirmed brain amyloid-β pathology to establish the detailed efficacy of therapeutic plasma exchange and albumin supplement in AD. On the flip side, there is a gap in our scientific understanding of the Aβ dynamics between the brain, plasma, and CSF, which can affect the development of such therapeutic strategies.
The advent of anti-amyloid therapies in AD certainly presents a challenge for enrolling participants in plasma exchange trials. These therapies have shown promise, particularly in cases of MCI or early dementia. However, this should not deter the exploration of other potential treatments. If plasma exchange with albumin replacement proves beneficial in more extensive trials, it could significantly expand the range of therapeutic options available for AD. Diversifying treatment approaches is crucial, as it provides multiple avenues to combat the disease and potentially improve patient outcomes. Thus, continuing to investigate plasma exchange is important, despite the current focus on anti-amyloid therapies.
Funding sourcesNil.
Conflict of interestDr. Ritwick Mondal (ritwickraw@gmail.com) reports no relevant disclosures.
Dr. Shramana Deb (shramanadeb1995@gmail.com) reports no relevant disclosures.
Dr. Gourav Shome (gshome007@gmail.com) reports no relevant disclosures.
Dr. Vramanti Sarkar (vramantisarkar@gmail.com) reports no relevant disclosures.
Dr. Durjoy Lahiri (dlahiri1988@gmail.com) reports no relevant disclosures.
Dr. Suvro Sankha Datta (suvro.datta@gmail.com) reports no relevant disclosures.
Dr. Julián Benito-León (jbenitol67@gmail.com) reports no relevant disclosures.
None of us has conflict of interests.
J. Benito-León is supported by the National Institutes of Health, Bethesda, MD, USA (NINDS #R01 NS39422), the European Commission (grant ICT-2011-287739, NeuroTREMOR), the Spanish Health Research Agency (grant FIS PI12/01602 and grant FIS PI16/00451) and The Recovery, Transformation, and Resilience Plan at the Ministry of Science and Innovation (grant TED2021-130174B-C33, NETremor).

