



La epilepsia con mioclonías palpebrales es una epilepsia generalizada idiopática infantil que está infradiagnosticada, ya que se confunde con tics o una acción conductual.
Pacientes y métodosEstudio cuantitativo, observacional, descriptivo de 16 pacientes con epilepsia y mioclónicas palpebrales atendidos en una institución especializada en neurología, entre los años 2017 y 2022. Se evaluaron las historias clínicas y los videoelectroencefalogramas.
ResultadosDe 16 pacientes, 11 eran mujeres (68,8%), la mediana de edad fue de 17,5 años (RIC 12,5). El primer diagnóstico que los pacientes recibieron con mayor frecuencia fue epilepsia idiopática generalizada. Las situaciones estresantes fueron el precipitante más reportado. El 93,75% de los pacientes presentaron inicialmente mioclonías palpebrales y al cabo de varios años presentaron convulsiones tónico-clónicas generalizadas. El tiempo transcurrido entre la primera convulsión y el diagnóstico varió entre 1 y más de 40 años, fue mayor entre los pacientes con sistema de salud subsidiado. Los hombres tenían una alta frecuencia de antecedentes familiares de epilepsia en primer y segundo grado de consanguinidad, presentaron mayor demora en el diagnóstico y reportaron mayor farmacorresistencia. Tanto en mujeres como en hombres las regiones posteriores (occipital, temporoccipital y parietoccipital) presentaron mayor actividad epileptiforme focal. La respuesta fotoparoxística se presentó con mayor predominio en las mujeres; las respuesta del tipo iii y iv de Waltz fueron las más frecuentes.
ConclusiónSe debe resaltar la importancia de sospechar esta enfermedad ante mioclonías palpebrales en población infantil, garantizando calidad y manejo oportunos.
Epilepsy with palpebral myoclonus is an idiopathic generalized childhood epilepsy, which is underdiagnosed because it is confused with tics or behavioral actions.
Patients and methodsQuantitative, observational, descriptive of 16 patients with epilepsy and myoclonic eyelids treated at an institution specialized in neurology, between the years 2017 and 2022. Clinical histories and videoelectroencephalograms were evaluated.
ResultsOf 16 patients, 11 were women (68.8%), the median age was 17.5 years (IQR 12.5). The first diagnosis most frequently received by patients was idiopathic generalized epilepsy. Stressful situations were the most reported precipitant. 93.75% of the patients initially presented palpebral myoclonic seizures and after several years presented generalized tonic-clonic seizures. The time elapsed between the first seizure and the diagnosis varied between 1 and more than 40 years, being greater among patients with a subsidized health system. Men had a high frequency of family history of epilepsy in the first and second degrees of consanguinity, had a longer delay in diagnosis and reported greater drug resistance. In both women and men, the posterior regions (occipital, temporoccipital and parietoccipital) presented a greater focal epileptiform activity. The photoparoxysmal response occurred with greater predominance in women, with the Waltz type iii and iv responses being the most frequent.
ConclusionThe importance of suspecting this pathology before palpebral myoclonus in children should be highlighted, guaranteeing quality and timely management.
La epilepsia con mioclonías palpebrales (EMP), nombrada anteriormente como síndrome de Jeavons (SJ), es una epilepsia generalizada idiopática infantil que comparte elementos electroclínicos con otras epilepsias idiopáticas o genéticas generalizadas. Este síndrome no cuenta con datos epidemiológicos exactos, debido a las dificultades diagnósticas que representa y a la falta de reconocimiento del personal médico. Se calcula que la EMP representa del 2,7% al 12,9% de las epilepsias generalizadas1, con una prevalencia del 3% en adultos y del 13% en la población infantil con epilepsia generalizada idiopática con ausencias2 y representa del 0,56% al 2,7% de todas las epilepsias3. La EMP se presenta en la infancia entre los 2 y 14 años (media 6,5±2,5 años)4 y predomina en el sexo femenino con una relación de mujer a hombre de 2:11,5. Se presume que la EMP es una enfermedad tan frecuente como la epilepsia mioclónica juvenil, sin embargo, es infradiagnosticada porque se confunde y diagnostica como tics o una acción conductual3,4.
La EMP se caracteriza por la siguiente tríada: mioclonías palpebrales con o sin ausencia6, crisis con un patrón EEG con breve actividad bilateral de polipunta-onda generalizada de 3 a 6 Hz inducida por el cierre ocular en ambientes luminosos3 y fotosensibilidad o respuesta fotoparoxística6, que tiende a disminuir con el paso del tiempo.
La etiología de este síndrome es aún motivo de estudio. Se ha propuesto un carácter genético de acuerdo con estudios de casos de gemelos monocigóticos concordantes3. Las mutaciones que se han relacionado con esta enfermedad son la mutación KCNB1, KIAA2022, NAA10 y variaciones en la CHD2 (proteína de unión al ADN helicaso de dominio cromo 2)6. En la EMP con ausencias se han descrito 4 genes con posible asociación (SYNGAP1, KIA02022/NEXMIF, RORB y CHD2) y se han reportado en algunos pacientes diferentes alteraciones en 3 genes (SLC2A1, NAA10 y KCNB1), aunque falta mayor evidencia para establecer una relación más clara7. Alrededor del 20% de los pacientes con EMP presentan antecedentes familiares positivos5. Se cree que se presenta un mal funcionamiento del generador de ritmo α en la corteza occipital6 y se ha propuesto la posibilidad de que esta corteza inicie una red de epilepsia generalizada que incluya el tronco encefálico, las vías tálamocorticales y transcorticales8,9.
El tratamiento más utilizado es el ácido valproico; sin embargo, por su efecto teratogénico se evita en mujeres en edad reproductiva y con deseos de quedar embarazadas4,6: en esta población se prefiere el uso de levetiracetam. Otros medicamentos útiles son la lamotrigina, etosuximida, zonisamida, que pueden administrarse en monoterapia o en combinación con el ácido valproico2,10.
Los pacientes con EMP tienden a ser farmacorresistentes, por lo que requieren de la combinación de múltiples medicamentos asociado a otras alternativas terapéuticas2,10,11.
Colombia no cuenta con datos epidemiológicos de la EMP, tampoco existen registros estadísticos claros sobre datos clínicos, características sociodemográficas, electroencefalográficas, respuesta terapéutica, comorbilidades ni formas en las que se presenta el síndrome, al igual que el tiempo en que tarda el diagnóstico o el inicio del tratamiento. Por ende, el objetivo de este estudio fue realizar una descripción de las características sociodemográficas y clínicas de una serie de casos de pacientes colombianos con esta enfermedad.
Material y métodosEstudio cuantitativo, observacional, descriptivo de 16 pacientes con epilepsia y mioclónicas palpebrales atendidos en el Fundación Instituto Neurológico de Colombia (FINDEC) entre los años 2017 y 2022. La fuente de información documental fueron la historia clínica y el videoelectroencefalograma de cada paciente. Se incluyeron todos los pacientes con diagnóstico confirmado de EMP o SJ y se descartaron a los que no contaran con registro de la videoelectroencefalografía.
Se describen, según el sexo, las variables cuantitativas con medianas y rango intercuartílico y las variables cualitativas por medio de frecuencias absolutas y relativas. Se uso el programa de acceso libre Jamovi 2.2.5.
Este fue un estudio aprobado por Comité Deética de la institución hospitalaria en el acta de reunión número 128. Se conservó el anonimato y confidencialidad durante todas las etapas de la investigación.
ResultadosEntre los años 2017 y 2022 se identificaron 16 pacientes con diagnóstico confirmado de EMP. El sexo femenino representó el 68,8% (11/16) y para el año 2022 los pacientes se encontraban entre los 9 y los 54 años (mediana de 17,5; IQR 12,5).
La mayoría de los pacientes residían y procedían de los municipios del Valle de Aburra (9), un paciente era originario de Venezuela. La conformación familiar más frecuente fue tipo nuclear y extensa.
Entre los antecedentes perinatales, 2pacientes nacieron en casa sin complicaciones. Un paciente nació pretérmino con retraso del crecimiento intrauterino y dificultad respiratoria; otro paciente fue producto de un parto prolongado y distócico; otro sujeto nació con microcefalia y desarrolló ictericia neonatal.
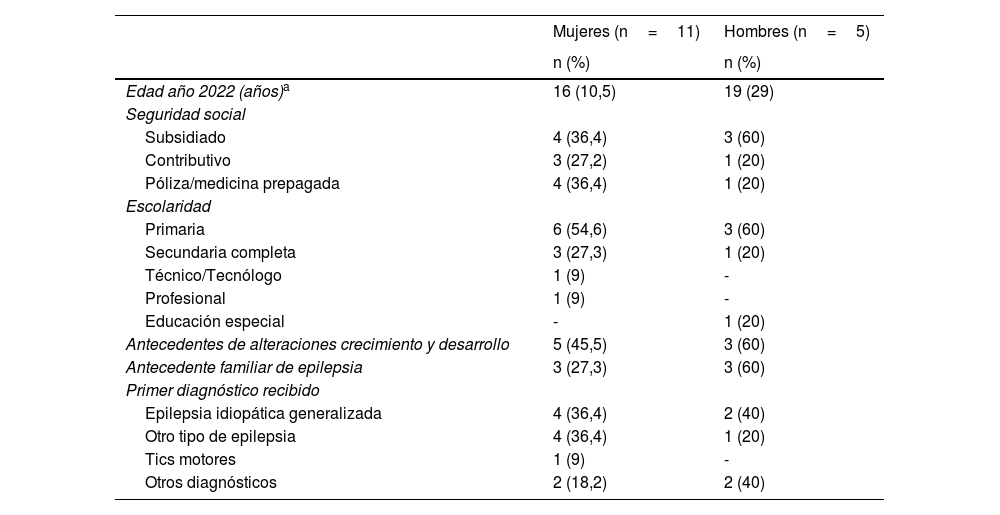
El régimen de seguridad social en salud de tipo contributivo y complementario fue el más frecuente (63,6%) entre las mujeres, mientras que el subsidiado lo fue entre los hombres. Mas del 80% de los pacientes habían finalizado estudios de primaria o secundaria (tabla 1).
Caracterización sociodemográfica y clínica según sexo
| Mujeres (n=11) | Hombres (n=5) | |
|---|---|---|
| n (%) | n (%) | |
| Edad año 2022 (años)a | 16 (10,5) | 19 (29) |
| Seguridad social | ||
| Subsidiado | 4 (36,4) | 3 (60) |
| Contributivo | 3 (27,2) | 1 (20) |
| Póliza/medicina prepagada | 4 (36,4) | 1 (20) |
| Escolaridad | ||
| Primaria | 6 (54,6) | 3 (60) |
| Secundaria completa | 3 (27,3) | 1 (20) |
| Técnico/Tecnólogo | 1 (9) | - |
| Profesional | 1 (9) | - |
| Educación especial | - | 1 (20) |
| Antecedentes de alteraciones crecimiento y desarrollo | 5 (45,5) | 3 (60) |
| Antecedente familiar de epilepsia | 3 (27,3) | 3 (60) |
| Primer diagnóstico recibido | ||
| Epilepsia idiopática generalizada | 4 (36,4) | 2 (40) |
| Otro tipo de epilepsia | 4 (36,4) | 1 (20) |
| Tics motores | 1 (9) | - |
| Otros diagnósticos | 2 (18,2) | 2 (40) |
De los 16 pacientes, 8tuvieron alteraciones en el crecimiento y desarrollo, con el desarrollo cognitivo como el más frecuentemente alterado 75% (6/8) y el compromiso del lenguaje como el más reportado (62,5%). Antes del inicio de los cuadros convulsivos no se reportó antecedente de trauma craneoencefálico. Entre los antecedentes personales de importancia se destacan: acidosis tubular renal en una mujer, un hombre monorrenal y 2pacientes con parálisis cerebral infantil. Dos pacientes tenían estudio de cariotipo y solo uno tenía alteraciones; hubo una paciente con cromosomopatía por duplicación del material genético de origen desconocido 47XX+ mar de origen del gen 18p11.32p11.21.
Los hombres tenían una alta frecuencia de antecedentes familiares de epilepsia en primer y segundo grado de consanguinidad. El primer diagnóstico que los pacientes recibieron con mayor frecuencia fue epilepsia idiopática generalizada (2 hombres y 4 mujeres), entre los otros diagnósticos recibidos estuvieron convulsiones febriles, episodios paroxísticos y ceguera cortical (tabla 1).
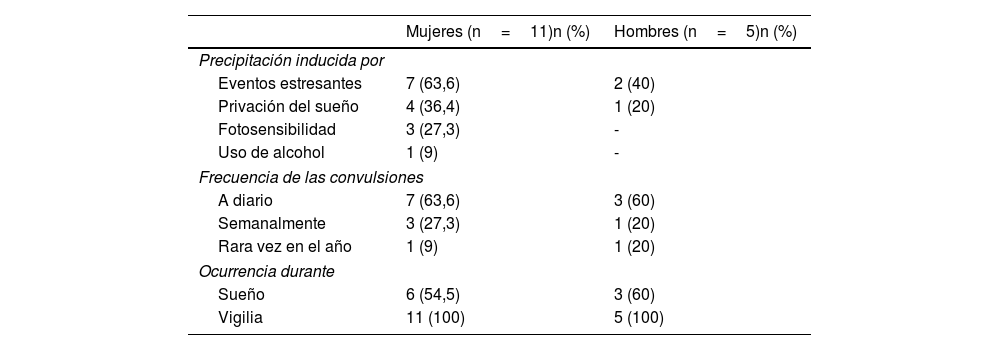
La descripción semiológica de las convulsiones corresponde a las características reportadas en la historia clínica antes de recibir el diagnóstico de EMP. El precipitante más identificado por los pacientes fueron las situaciones estresantes. Según las frecuencias de las convulsiones lo más usual era la presentación a diario en cualquier hora del día. Solo un paciente hombre reportó convulsiones diarias en horas de la mañana. En los registros médicos se reportó el reconocimiento de convulsiones durante los periodos de vigilia en el 100% de los pacientes y en más del 50% durante el periodo de sueño (tabla 2).
Características semiológicas de los episodios convulsivos según sexo
| Mujeres (n=11)n (%) | Hombres (n=5)n (%) | |
|---|---|---|
| Precipitación inducida por | ||
| Eventos estresantes | 7 (63,6) | 2 (40) |
| Privación del sueño | 4 (36,4) | 1 (20) |
| Fotosensibilidad | 3 (27,3) | - |
| Uso de alcohol | 1 (9) | - |
| Frecuencia de las convulsiones | ||
| A diario | 7 (63,6) | 3 (60) |
| Semanalmente | 3 (27,3) | 1 (20) |
| Rara vez en el año | 1 (9) | 1 (20) |
| Ocurrencia durante | ||
| Sueño | 6 (54,5) | 3 (60) |
| Vigilia | 11 (100) | 5 (100) |
| Mediana (RIC) | Mín-Máx | Mediana (RIC) | Mín-Máx | |
|---|---|---|---|---|
| Edad de la primera crisis mioclónica palpebral (años) a | 6 (3) | 0,3-11 | 5 (2) | 2-9 |
| Edad de la primera crisis TCG (años) a | 9 (2,5) | 8-13 | 9 (14) | 3-41 |
| Edad en la que se establece el diagnóstico | 10 (7) | 5-46 | 8 (24) | 7-46 |
| Tiempo al diagnóstico (años)b | 5 (7) | 1-40 | 6 (11) | 2-42 |
| Régimen Sistema de Salud | N (%) | Mediana (RIC) | Mín-Máx | N (%) | Mediana (RIC) | Mín-Máx |
|---|---|---|---|---|---|---|
| Subsidiado | 4 (36,4) | 7 (13) | 3-40 | 3 (60) | 6 (18) | 5-42 |
| Contributivo | 3 (27,2) | 2 (2) | 1-5 | 1 (20) | 2 (0) | - |
| Póliza/prepagada | 4 (36,4) | 9 (6) | 5-14 | 1 (20) | 32 (0) | - |
| Brecha terapéutica (meses) c | - | 2 (2) | 0-6 | - | 7 (2) | 5-9 |
TCG: tónico-clónica generalizada.
En el 93,75% de los pacientes se reportaron inicialmente convulsiones mioclónicas palpebrales y al cabo de varios años presentaron convulsiones tónico-clónicas generalizadas.
La presentación de la primera convulsión mioclónica palpebral se reportó principalmente en la infancia; solo 2pacientes la presentaron antes del año de vida. Los años transcurridos entre la primera convulsión y el diagnóstico de EMP varió entre 1 y más de 40 (mediana 5,5 años; RIC 8), tanto en el grupo de hombres como en las mujeres. Al analizar según seguridad social, los pacientes del régimen subsidiado fueron los que más retraso en el diagnóstico presentaron. El tiempo transcurrido entre el diagnóstico por videoelectroencefalograma y el inicio del tratamiento específico fue variado, aunque en todos los casos menor a 10 meses (tabla 2).
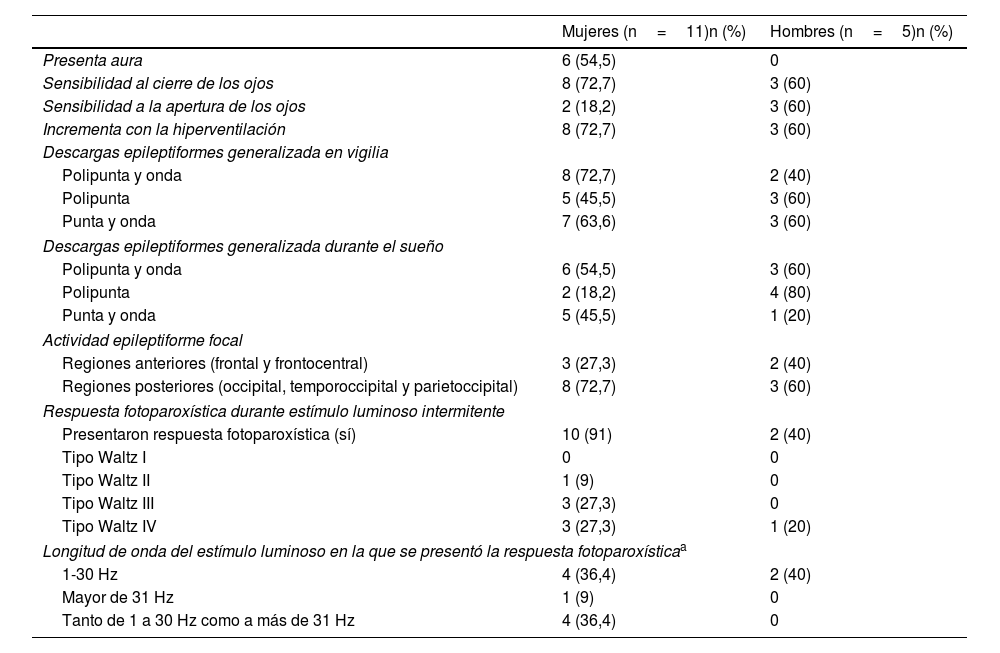
Al videoelectroencefalograma se evidenció que más del 50% de las mujeres presentaban aura, no así en los hombres. Además, en las mujeres las mioclonías de los párpados fueron más frecuentes al cierre de los ojos (72%) que a la apertura (18%). Con la hiperventilación se presentó incremento de las crisis en más de la mitad de los participantes. Durante la vigilia y el sueño el patrón de descargas de epilepsia generalizada en mujeres más frecuente fue polipunta y onda, mientras que en los hombres fue polipunta y punta-onda durante la vigilia y polipunta durante el sueño. Tanto en mujeres como en hombres las regiones posteriores (occipital, temporoccipital y parietoccipital) presentaron con mayor frecuencia actividad epileptiforme focal (tabla 3).
Características al videoelectroencefalograma
| Mujeres (n=11)n (%) | Hombres (n=5)n (%) | |
|---|---|---|
| Presenta aura | 6 (54,5) | 0 |
| Sensibilidad al cierre de los ojos | 8 (72,7) | 3 (60) |
| Sensibilidad a la apertura de los ojos | 2 (18,2) | 3 (60) |
| Incrementa con la hiperventilación | 8 (72,7) | 3 (60) |
| Descargas epileptiformes generalizada en vigilia | ||
| Polipunta y onda | 8 (72,7) | 2 (40) |
| Polipunta | 5 (45,5) | 3 (60) |
| Punta y onda | 7 (63,6) | 3 (60) |
| Descargas epileptiformes generalizada durante el sueño | ||
| Polipunta y onda | 6 (54,5) | 3 (60) |
| Polipunta | 2 (18,2) | 4 (80) |
| Punta y onda | 5 (45,5) | 1 (20) |
| Actividad epileptiforme focal | ||
| Regiones anteriores (frontal y frontocentral) | 3 (27,3) | 2 (40) |
| Regiones posteriores (occipital, temporoccipital y parietoccipital) | 8 (72,7) | 3 (60) |
| Respuesta fotoparoxística durante estímulo luminoso intermitente | ||
| Presentaron respuesta fotoparoxística (sí) | 10 (91) | 2 (40) |
| Tipo Waltz I | 0 | 0 |
| Tipo Waltz II | 1 (9) | 0 |
| Tipo Waltz III | 3 (27,3) | 0 |
| Tipo Waltz IV | 3 (27,3) | 1 (20) |
| Longitud de onda del estímulo luminoso en la que se presentó la respuesta fotoparoxísticaa | ||
| 1-30 Hz | 4 (36,4) | 2 (40) |
| Mayor de 31 Hz | 1 (9) | 0 |
| Tanto de 1 a 30 Hz como a más de 31 Hz | 4 (36,4) | 0 |
La respuesta fotoparoxística durante el estímulo luminoso intermitente se presentó con mayor predominio en las mujeres: la respuesta de tipo iii (puntas y ondas parietoccipital que se extienden a la región frontal) y la iv (puntas y ondas generalizadas) de Waltz fueron las más frecuentes12. La longitud de onda que más desencadenó respuesta fotoparoxística fue de 1 a 30Hz (tabla 3).
Algunos pacientes fueron estudiados con otras pruebas, como: neuroimágenes (TAC 2, RMN 11, ambos 1), polisomnografía (1), potenciales evocados auditivos (1) y visuales (2). Tres pacientes presentaron alteraciones en estas ayudas diagnósticas.
La mayoría de las neuroimágenes fueron normales. Solo un paciente con antecedente de parálisis cerebral infantil evidenció en la RMN encefalopatía hipóxico-isquémica, degeneración walleriana y esclerosis hipocampal derecha. En otro paciente se reportó, en el potencial evocado auditivo, hipoacusia leve en el oído derecho y un potencial evocado visual anormal en el lado derecho por latencia y por ausencia en el lado izquierdo, en cuya polisomnografía se evidenció ausencia de REM, apnea, hipopnea, EEG anormal punta-onda lenta y polipunta generalizada. Otro paciente presentó en los potenciales evocados visuales ausencia completa de la respuesta cortical al estímulo de flash y patrón de flash de ambos ojos.
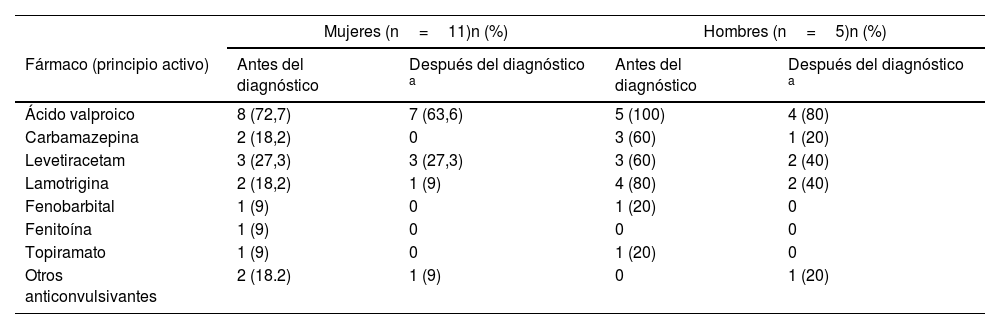
El ácido valproico fue el medicamento más formulado tanto antes del diagnóstico como después. El segundo medicamento más prescrito antes de la confirmación del diagnóstico en mujeres fue levetiracetam y en hombres fue lamotrigina. Después de la confirmación del diagnóstico, el ácido valproico fue el más formulado en ambos sexos. La monoterapia fue más habitual en mujeres, con la que logró el control en la mayoría de los casos. En hombres se ordenó principalmente la combinación de 2medicamentos y se reportó control parcial con mayor farmacorresistencia (tabla 4).
Características de la respuesta terapéutica
| Mujeres (n=11)n (%) | Hombres (n=5)n (%) | |||
|---|---|---|---|---|
| Fármaco (principio activo) | Antes del diagnóstico | Después del diagnóstico a | Antes del diagnóstico | Después del diagnóstico a |
| Ácido valproico | 8 (72,7) | 7 (63,6) | 5 (100) | 4 (80) |
| Carbamazepina | 2 (18,2) | 0 | 3 (60) | 1 (20) |
| Levetiracetam | 3 (27,3) | 3 (27,3) | 3 (60) | 2 (40) |
| Lamotrigina | 2 (18,2) | 1 (9) | 4 (80) | 2 (40) |
| Fenobarbital | 1 (9) | 0 | 1 (20) | 0 |
| Fenitoína | 1 (9) | 0 | 0 | 0 |
| Topiramato | 1 (9) | 0 | 1 (20) | 0 |
| Otros anticonvulsivantes | 2 (18.2) | 1 (9) | 0 | 1 (20) |
| Mujeres (n=11)n (%) | Hombres (n=5)n (%) | |
|---|---|---|
| Monoterapia | 9 (81,8) | 1(20) |
| 2 medicamentos | 1 (9) | 4 (80) |
| No recibe anticonvulsivantes | 1 (9) | 0 |
| Control con los medicamentos actuales | ||
| Sí | 4 (36,4) | 1 (20) |
| No | 0 | 1 (20) |
| Parcialmente | 2 (18,2) | 2 (40) |
| No se reporta | 5 (45,4) | 1 (20) |
| Continuidad en el tratamiento reportado en la HC | ||
| Sí | 3 (27,3) | 3 (60) |
| No, por falta de suministro por la EPS | 0 | 1 (20) |
| No, por decisión del paciente o acudiente | 1 (9) | 1 (20) |
| No se reporta | 7 (63,6) | 0 |
| Farmacorresistenciab | 2 (18,2) | 3 (60) |
EPS: entidad promotora de salud; HC: historia clínica.
Las terapias no farmacológicas (lentes blue Z1, estimulación del nervio vago, neuroestimulación receptiva del tálamo) no tienen registro en la historia clínica de los pacientes. Se reportó el caso de un paciente tratado con dieta cetogénica que alcanzó control adecuado de las crisis y el de otra paciente tratada con cannabidiol, que logró un control adecuado de las descargas epileptiformes.
DiscusiónLos resultados del presente estudio permiten hacer un análisis en 2ejes. El primero de ellos evidencia las similitudes con lo reportado en la literatura sobre EMP y el segundo, relacionado con el efecto que tiene el sistema de salud en Colombia en los pacientes con esta enfermedad. Ambos ejes de análisis permiten comprender de forma general, a nivel nacional y de forma específica en la ciudad de Medellín, las características del síndrome y las implicaciones del contexto socioeconómico de los pacientes al momento del diagnóstico y tratamiento.
En esta investigación se describen los hallazgos sociodemográficos y clínicos a partir de las historias clínicas y videoelectroencefalogramas de 16 pacientes con EMP atendidos en una institución especializada en neurología de la ciudad de Medellín. Por las características específicas de las convulsiones, los hallazgos electroclínicos y la resistencia al tratamiento, la EMP es considerada una epilepsia poco reconocida o que tiende a ser errónea y tardíamente diagnosticada, lo cual hace difícil estimar la prevalencia exacta13-15. En la bibliografía se ha descrito que representa del 2,7% al 12,9% de las epilepsias generalizadas y del 0,56% al 2,7% de todas las epilepsias1,16,17. Los hallazgos aquí encontrados, en cuanto edad y sexo, son similares a lo reportado por otros autores: la edad de inicio típicamente ocurre en la niñez4 y hay predominio en el sexo femenino3,13. Sin embargo, existen otros estudios en los cuales se reporta que la proporción de sexos era casi igual14,15. Esta distribución sexual desigual puede deberse a los diferentes antecedentes genéticos de las diversas poblaciones15. En cuanto a la edad, cabe resaltar que 2de los pacientes presentaron EMP antes del año de vida. En la bibliografía se han descrito subtipos clínicos de esta epilepsia que, para el caso de estos 2pacientes, correspondería a una epilepsia de aparición temprana con mioclonías palpebrales4,6.
La mitad de los pacientes reportaron antecedentes familiares de epilepsia y, de estos, el 37,5% tenían familiares en primer y segundo grado de consanguinidad. Este resultado concuerda con lo expresado en diversos estudios, en los cuales los antecedentes familiares de epilepsia en EMP se informan en tasas entre el 33% y el 83%15,18,19.
En la EMP se han descrito otros tipos de convulsiones además de las mioclonías palpebrales. En nuestra serie de casos el 43,75% presentó por lo menos una vez en la vida convulsiones tónico-clónicas generalizadas, las cuales han sido reportadas en más de la mitad de los pacientes y suelen ser inevitables a largo plazo13,20.
La respuesta fotoparoxística se informa en todos los pacientes jóvenes sin tratamiento, que puede reducirse con los años o por la terapia antiepiléptica5,21,22. En este estudio se reportó en el 75% de los pacientes, principalmente en el sexo femenino.
Los precipitantes de las convulsiones más identificados por los pacientes fueron las situaciones estresantes (10 pacientes), seguido de privación del sueño (5 pacientes), suspensión del medicamento (3 pacientes), exposición a luces intermitentes o uso de dispositivos electrónicos (3 pacientes), consumo de licor, entre otros; datos similares a los descritos por Pérez-Errazquin et al.23. Dos de los pacientes reportaron desencadenantes diferentes a los encontrados en la bibliografía. Una de las pacientes identificó un incremento de los episodios con el periodo menstrual, mientras que otra lo asoció al ayuno prolongado. Los datos mencionados permiten inferir que los precipitantes son variados y diferentes en cada paciente. El reconocimiento de estos desencadenantes durante la atención medica es fundamental para intervenir o evitar el aumento de las crisis y así mejorar la calidad de vida de los pacientes.
El 93,75% de los pacientes tenían reporte de neuroimagen normal, hallazgo similar a lo descrito en la bibliografía, en la que las RM son normales o no específicas en esta población3,6.
En relación con el desempeño académico de los pacientes del presente estudio, fue reportado como regular en el 80%, con repitencia académica en el 33,3% y se describieron principalmente dificultades en la adquisición del conocimiento, inatención y dificultades para seguir instrucciones. También se ha reseñado un funcionamiento neurocognitivo con tendencia a estar en el límite inferior sin ser catalogado como discapacidad intelectual14,24,25. El rendimiento académico muestra tendencia a la baja, sin embargo, no existen estudios que determinen si esta alteración en el aprendizaje es inherente al síndrome, debido a que puede asociarse a las múltiples descargas epileptiformes o ser consecuencia del uso o mala prescripción de medicamentos anticrisis24.
El tiempo para que los pacientes fueran diagnosticados con EMP fue en promedio de 11,64 años desde que el primer episodio. Este tiempo es mayor que lo especificado por Reyhani y Özkara15, para quienes el intervalo fue de 9 años, o lo descrito por otros autores, quienes informaron un retraso de 10 años o menos3,26. En la mayoría de los pacientes el primer diagnóstico estuvo asociado a algún tipo de epilepsia, especialmente la idiopática generalizada y las epilepsias focales. Otros diagnósticos fueron tics, ceguera cortical, convulsiones febriles, TDAH, entre otros. La EMP aún no es bien conocida ni reconocida por el personal médico y tiende a ser confundida con otros tipos de trastornos, lo cual conlleva el retraso en el diagnóstico15,27.
El diagnóstico inadecuado de los pacientes resulta en tratamientos contraindicados. De los 16 pacientes, 5 fueron tratados con carbamazepina y oxcarbamazepina. El uso de estos medicamentos no es recomendado en este tipo de epilepsia por presentar empeoramiento de las crisis6, además de los efetos secundarios que pueden llegar a presentarse, lo que puede condicionar el abandono del tratamiento.
Al tener en cuenta el régimen del sistema de seguridad social y el tiempo en años que tardó el diagnóstico, se identificó que los pacientes con un retraso mayor a 5 años pertenecían al régimen subsidiado. Es bien conocido el hecho de que las necesidades más básicas en salud de los países en vía de desarrollo siguen sin satisfacerse y que la situación empeora cuando se requiere atención de alta calidad que incluya personal sanitario y equipos médicos especializados28, como es el caso de la EMP. Además del reto que implica el diagnóstico oportuno de esta enfermedad, en nuestro caso se suman las barreras que surgen para el acceso a la atención médica. Aunque el Sistema de Seguridad Social en Salud en Colombia incrementó la cobertura de la población, también llevo a un aumento en la demanda de servicios médicos, incluido el recurso humano y equipos especializados29. Además, la inequidad entre regiones lleva a la falta de acceso a los servicios de salud y a la escasez de personal médico especialista30.
La mayoría de los pacientes de este estudio presentaron en las regiones posteriores (occipital, temporoccipital y parietoccipital) mayor actividad epileptiforme focal, de forma similar a lo descrito por Nilo et al.27 en el 60% de los casos de su cohorte. Estos resultados son explicados por los hallazgos de neuroimagen funcional, que han mostrado un estado de hiperexcitabilidad de la corteza occipital en pacientes con EMP31. Esta actividad permanece limitada a esta región o puede incluir el tronco encefálico, activando las redes talamocorticales y transcorticales causantes de las convulsiones generalizadas8.
Valorar la farmacorresistencia en todos los pacientes a partir de las historias clínicas no fue posible, debido a la falta de documentación de esta variable en algunos de los registros. Sin embargo, se observó que entre los hombres se presentó mayor combinación de medicamentos y menor control de las crisis. En la bibliografía está descrito que la EMP es un trastorno de por vida, que suele ser muy resistente al tratamiento y que la mioclonía palpebral suele persistir hasta la edad adulta32. Para Smith et al.3, las convulsiones tónico-clónicas generalizadas y los tipos de convulsiones distintos de las crisis de ausencia pueden ser factores predictores de epilepsia resistente a los medicamentos. Zawar y Pestana5 refieren que los hombres tiene un mejor pronóstico que las mujeres, al contrario de lo observado en este estudio; sin embargo, se debe tener en cuenta que el número de pacientes hombres en esta serie fue pequeño y que puede ser un hallazgo incidental.
En cuanto al uso de otras terapias no farmacológicas, se encontró en un paciente la instauración de una dieta cetogénica. Este tipo de terapia dietética es una alternativa en pacientes con epilepsias refractarias al tratamiento. Ruiz Herrero et al.33 realizaron un seguimiento de 18 años a 160 pacientes pediátricos epilépticos tratados con dieta cetogénica y reportaron que el 12-15% quedó libre de crisis y, según el tiempo de seguimiento, una buena respuesta a los 3 meses del 41,9% y a los 24 meses del 16,2%. Sin embargo, los efectos adversos son frecuentes y pueden afectar al estado nutricional y al crecimiento. En trabajos previos el 15% de los pacientes quedaban libres de crisis y más del 30% presentaban una reducción superior al 50%34-36. En la bibliografía no existen estudios específicos para EMP. Otra de las terapias establecidas para uno de los pacientes de esta serie fue el cannabidiol. Se Hee et al.37 refieren que es un fármaco anticonvulsivo eficaz que se utiliza para tratar el síndrome de Dravet y el síndrome de Lennox-Gastaut, sin embargo, no mejoró la calidad de vida de los 41 pacientes evaluados. Por otro lado, Zawar et al.38 describieron el uso de cannabidiol en 2pacientes con EMP y reportaron exacerbación de las mioclonías palpebrales, las cuales cesaron al retirarlo. Se requieren más estudios en esta población para esclarecer la efectividad de este químico.
Entre las limitaciones se tiene que este estudio fue de naturaleza retrospectiva y se basó en notas clínicas y documentación. Algunas variables no estaban reportadas y el seguimiento de la eficacia de los medicamentos no se documentó sistemáticamente en todos los casos.
ConclusionesLa EMP o SJ es un trastorno convulsivo que se observa en la infancia y tiene síntomas de por vida. Se debe resaltar la importancia de sospechar esta enfermedad ante mioclonías palpebrales en población infantil con el fin de dar un diagnóstico oportuno y evitar el uso de medicamentos no recomendados para así mejorar la calidad de vida de esta población.
La comprensión de la fisiopatología de la enfermedad y el conocimiento de las características semiológicas y electroclínicas son importantes no solo para el neurólogo o epileptólogo, sino también para el médico general con el fin de que puedan realizar un abordaje inicial adecuado y remisión oportuna del paciente.
FinanciaciónLa Universidad Cooperativa de Colombia apoyó financieramente este estudio a través del Fondo de Investigación bajo el código de proyecto INV3177.
Conflicto de interesesLos autores declaran que no existe ningún conflicto de interés potencial con respecto a la investigación, autoría o publicación de este artículo.

