




La mucopolisacaridosis de tipo vi es una enfermedad lisosomal por déficit de arilsulfatasa B que conduce a una acumulación de dermatán sulfato. Los síntomas neurológicos incluyen compresión espinal, hidrocefalia y síndromes de atrapamiento. El diagnóstico se realiza por medio del dosaje de glucosaminoglucanos en orina y de la actividad enzimática en gota de sangre seca, leucocitos o cultivo de fibroblastos. Actualmente, la terapia de reemplazo enzimático (TRE) ha demostrado mejoría del compromiso esquelético, respiratorio y cardíaco. El compromiso medular no suele responder a la TRE cuando ya se encuentra presente, por lo que la descompresión quirúrgica debe ser indicada en forma temprana.
Mucopolysaccharidosis vi is a lysosomal storage disease due a deficiency of arylsulfatase B leading to the accumulation of dermatan sulfate. The disease shows a wide spectrum of symptoms from slowly to rapidly progressing forms. Neurological aspects are spinal compression, hydrocephalus and nerve entrapment. Diagnosis requires measurement of urinary glycosaminoglycans and arylsulfatase B activity in dried blood spot, leukocytes or cultured fibroblasts. Enzyme replacement therapy (ERT) showed improvement in skeletal, pulmonary and cardiac functioning. Spinal involvement does not respond to ERT when is present, surgical decompression should be indicated early.
La mucopolisacaridosis vi (MPS VI) o el síndrome de Maroteaux-Lamy es un trastorno autosómico recesivo resultado de la deficiencia o ausencia de la arilsulfatasa B (ASB)1, con la consecuente degradación incompleta y acumulación intralisosomal de glucosaminoglucanos (GAG), predominantemente dermatán sulfato (DS) y en mucho menor concentración condroitín sulfato en tejido conectivo2.
Los estudios epidemiológicos reportan tasas de prevalencia diversas desde 1 en 43.261 nacidos de inmigrantes turcos que viven en Alemania3 a 1 en 1.505.160 nacidos en Suecia4.
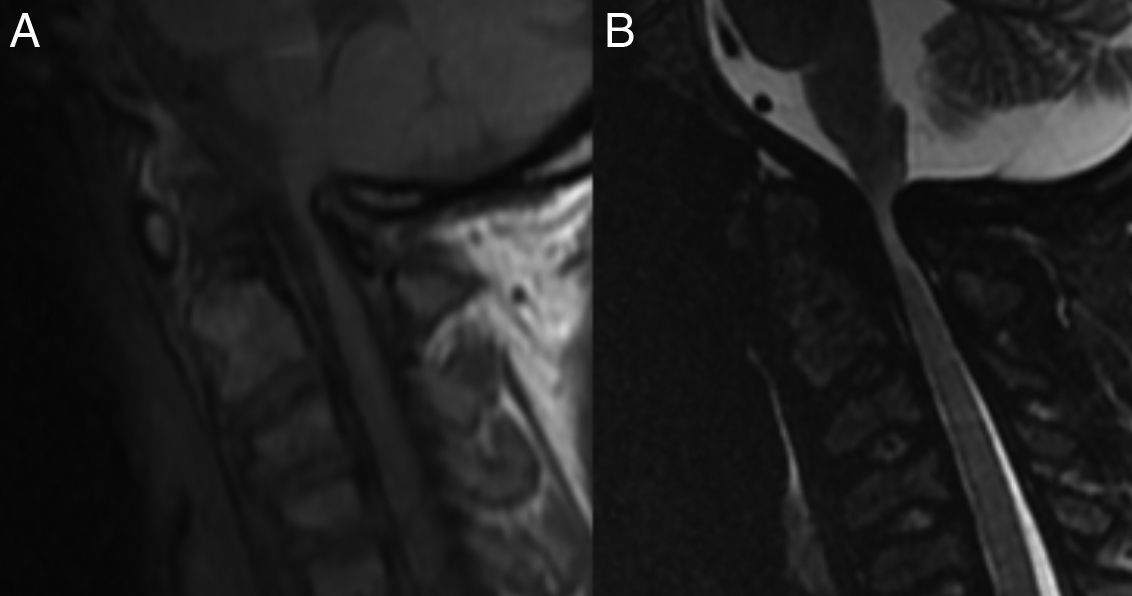
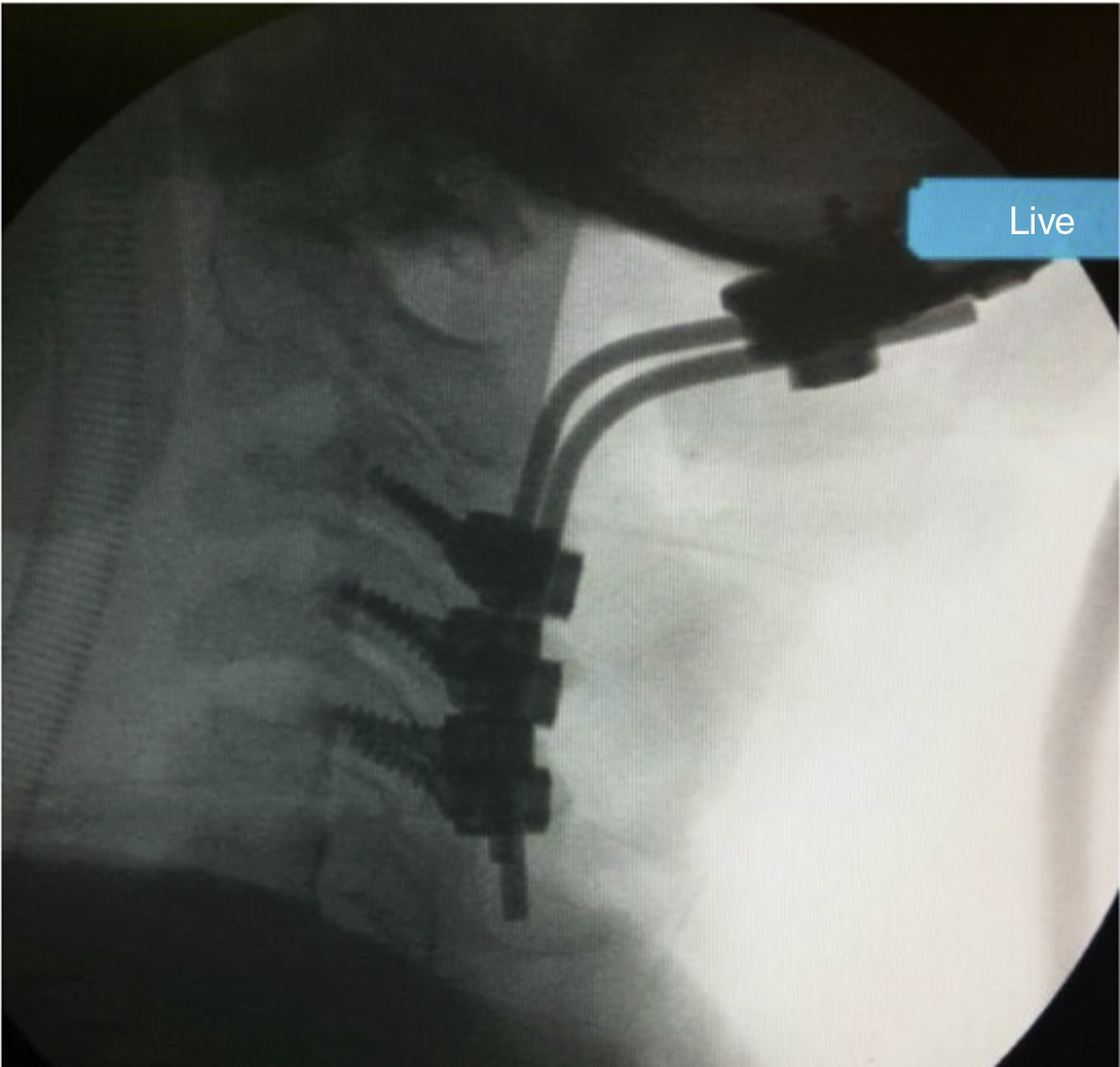
Caso clínicoSe presenta un paciente hombre de 26 años de edad, 145cm de altura, con diagnóstico confirmado de MPS vi a los 9 años, por medio de dosaje de ASB en leucocitos: 0,1 nmol/min/mg (VN 1,2 a 5,8) y niveles elevados de GAG en orina. El paciente había presentado displasia bilateral de cadera y hernia umbilical al nacimiento; al año se objetivó retraso en el crecimiento pondoestatural y a los 3 años, restricción para el movimiento articular a predominio en manos. Se realizó una amigdalectomía a los 11 años. Al iniciar la terapia de reemplazo enzimático (TRE), presentaba compromiso cervical medular, expresado por signos clínicos de liberación piramidal (clonus, signos de Hoffmann y Babinski) y daño medular cordonal posterior (dolor cervical focalizado, mioclonías propioespinales espontáneas y signo de Lhermitte). La resonancia de columna cervical mostraba severa compresión, con signos de mielomalacia a nivel C1-C2 (fig. 1). El ecocardiograma prequirúrgico mostró: insuficiencia aórtica moderada y estenosis mitral leve, el examen oftalmológico no informó compromiso corneano y la audiometría fue normal. Previo al inicio de la TRE, el test de marcha a los 6 min (6MWK) informó: 596 pasos (225 m) y el test de subida de escalones por 3 min (3MSC), 88 escalones. Luego de 10 meses de galsulafsa a 1mg/kg cada 14 días, no se evidenciaron reacciones alérgicas asociadas a las infusiones. El 6MWT se incrementó en 691 pasos (260 m) y el 3MSC, 98 escalones. La escala de calidad de vida (SF36) mostró clara mejoría, asociada a estabilidad en el resto de los estudios realizados. Si bien el beneficio de la TRE fue evidente, el dolor cervical persistió, asociado a un aumento de las mioclonías propioespinales, por lo que se realizó una cirugía descompresiva cervical con extracción del arco posterior de C1 y ampliación del foramen magno y posterior colocación prótesis con fijación superior occipital e inferior en C3 a C5 (fig. 2). Inmediatamente posterior a la recuperación anestésica, el paciente refirió alivio del dolor cervical y desaparición de las mioclonías propioespinales y del signo de Lhermitte. Continúa actualmente en TRE.
 Secuencia T1. B) Secuencia STIR. Compresión de la unión bulbo-cervical a nivel del foramen magno, con daño del cordón medular a ese nivel compatible con mielomalacia. Los cuerpos vertebrales muestran morfología atípica (signo de la «cabeza de pescado»).")

Luego de la evaluación de un grupo de 121 pacientes no tratados con MPS VI se establecieron 2 fenotipos: rápidamente y lentamente progresivos5.
La forma rápidamente progresiva se caracteriza por presentar niveles urinarios de GAG mayores a 200μcreatinina g/mg y no superar la estatura de 120cm. El inicio de los signos y síntomas suele ser antes de los 2 o 3 años de edad, con evidentes alteraciones esqueléticas, facies características y limitaciones para la movilidad articular predominantemente en manos (descritas como manos en flexión o en garra). Asociado a esto se evidencia retraso puberal, hepatoesplenomegalia, hernias umbilicales y/o inguinales, detención del crecimiento estatural con percentilos menores a 50 desde los 2 años de vida.1
El compromiso otorrinolaringológico implica trastornos en la audición, otitis media y sinusitis a repetición y síndrome de apnea obstructiva6. Las complicaciones pulmonares que se han descrito comparten características obstructivas y restrictivas7. La enfermedad pulmonar obstructiva es el resultado de la limitación del flujo aéreo y la presencia de traqueobroncomalacia, mientras que la enfermedad pulmonar restrictiva está dada por una caja torácica pequeña y rígida combinada con cifosis, escoliosis y lordosis lumbar aumentada.
Las complicaciones cardíacas, como estenosis o insuficiencia valvular, han sido descritas desde los 5 meses de vida. En frecuencia, la válvula mitral es la más afectada, seguida de la válvula tricúspide y finalmente la aórtica8.
El concepto de disostosis múltiple descrito característicamente en la MPS VI se relaciona a la presencia de macrocefalia, facies toscas con prominencia frontal exagerada, puente nasal deprimido, agrandamiento de la lengua, hipertrofia gingival y tardía erupción dental2. Los cuerpos vertebrales se describen como en «pico o cabeza de pescado», lo que favorece la cifosis dorso-lumbar.
Las complicaciones oftalmológicas más frecuentes son el glaucoma y la opacidad corneal, que pueden ser tratados con medicamentos y cirugía de trasplante de córnea, respectivamente. El compromiso del nervio óptico es secundario a la acumulación de GAG en la duramadre a nivel del foramen óptico y al subsecuente aumento de la presión intracraneal a ese nivel9.
Las complicaciones neurológicas se han dividido en centrales y periféricas. Estas últimas son el resultado de la compresión de los nervios mediano (túnel del carpo) y tibial posterior (túnel del tarso), que llevan frecuentemente a la indicación de cirugía de descompresión10.
Las complicaciones centrales son: la compresión bulbo-medular a nivel cervical (menos frecuente a nivel dorso-lumbar) y la hidrocefalia. Debe destacarse que la MPS VI no presenta compromiso intelectual ni cognitivo, como se ha descrito en otras MPS, como las enfermedades de Hurler (MPS I), de Hunter (MPS II) y de Sanfilippo (MPS III)11.
La compresión cervical ha sido descrita desde los 3 años de edad en pacientes con variante rápidamente progresiva y es el resultado del depósito de GAG en ligamentos transverso, cruzado y longitudinal anterior y posterior, tejidos blandos periodontoideos y duramadre. A estas anormalidades se suman la estenosis del foramen magno, con la consiguiente compresión de las arterias espinales, y la mielomalacia final, que resulta en cuadriparesia progresiva. La edad media de cirugía descompresiva en niños es de 12 años. El depósito de GAG en las vellosidades subaracnoideas puede limitar la reabsorción de líquido cefalorraquídeo, siendo causante de hidrocefalia comunicante, que requiere derivación ventrículo peritoneal temprana12.
La forma de presentación lentamente progresiva se caracteriza por alcanzar una altura mayor a 140cm y niveles de GAG urinarios por debajo de 100μcreatinina g/mg. Estos pacientes pueden desarrollar las mismas complicaciones esqueléticas que el fenotipo rápidamente progresivo pero en forma más atenuada. El diagnóstico ocurre generalmente después de los 5 años de edad y puede retrasarse hasta la segunda o tercera década de vida13.
El diagnóstico de MPS VI se realiza por medio del análisis de la actividad de la ASB en gota seca (técnica en papel de filtro), leucocitos aislados o cultivo de fibroblastos14. Siempre debe realizarse un dosaje de la actividad de otras sulfatasas, para excluir el diagnóstico de deficiencia de múltiples sulfatasas.
Se debe confirmar la presencia de GAG elevados en orina en primer término, para luego por medio de cromatografía de capa fina, identificar al DS como el GAG acumulado.
El test molecular para identificar mutaciones en el gen de la ASB no es mandatorio, aunque se recomienda su realización cuando los valores enzimáticos no son concluyentes14.
La TRE con galsulfasa fue aprobada por la Food and Drug Administration y Agencia Europea de Medicamentos para su comercialización y tratamiento de la MPS VI en el año 2005 y 2006, respectivamente. El primer estudio clínico controlado aleatorizado, doble ciego y multicéntrico, incluyó a 39 pacientes, donde los que recibieron galsulfasa luego de 24 semanas presentaron una mejoría significativa en el test 6MWK y el test 3MSC. Asociado a este beneficio, los valores de GAG urinarios disminuyeron en forma significativa y no hubo reacciones adversas serias15.
A la fecha se han publicado resultados de seguimiento de la función pulmonar y cardíaca16,17, ambos estudios demuestran resultados positivos con estabilización de la función sistólica, sin progresión del compromiso valvular y mejoría de la capacidad vital forzada y del volumen espiratorio forzado en muchos pacientes.
No hay evidencias de que la TRE mejore o detenga la progresión del daño medular espinal, cuando la compresión cervical ya está presente. Por el contrario, las distintas revisiones bibliográficas han mostrado progresión de los síntomas neurológicos durante la TRE, aun en presencia de mejoría cardíaca, motora y respiratoria, siendo la cirugía descompresiva la única intervención que puede mejorar los síntomas relacionados al compromiso espinal18.
ConclusionesEl paciente fue diagnosticado a los 9 años presentando un fenotipo lentamente progresivo. Como fue descripto anteriormente, este fenotipo evoluciona en forma más lenta, pero nunca debe hacer pensar a una variante «benigna», ya que habitualmente los pacientes requieren cirugía descompresiva cervical por daño isquémico medular, entre otras cirugías. Los resultados a 10 meses de TRE fueron positivos, con clara mejoría de la movilidad articular, incremento en el 6MWT en un 15% y del 3MSC del 11%. Aun así, se debió realizar una cirugía descompresiva cervical, lo que derivó en la inmediata disminución del dolor cervical y la desaparición de las mioclonías propioespinales y del signo de Lhermitte. Por lo descrito en la literatura y la evolución de nuestro paciente, se concluye que el tratamiento combinado entre TRE y cirugía debe ser indicado en forma temprana para un máximo beneficio en los pacientes.
