







La enfermedad de Pompe (EP) es un desorden metabólico autosómico recesivo infrecuente que se produce por ausencia o deficiencia de la enzima lisosomal alfa-glucosidasa ácida en los tejidos de los individuos afectados.
ObjetivoEl objetivo del presente consenso es revisar las pautas actuales y brindar recomendaciones para un correcto diagnóstico, evaluación, manejo y tratamiento de los pacientes con EP.
MétodosSe organizó un consenso que reunió profesionales nacionales y un invitado extranjero con experiencia en la EP en las áreas de clínica médica, clínica pediátrica, diagnóstico de laboratorio, neuropatología, neumonología, nutrición, neurología, enfermedades metabólicas, enfermedades neuromusculares (ENM) y rehabilitación de pacientes con ENM. Se realizó una revisión bibliográfica de las publicaciones y los artículos relevantes sobre EP existentes hasta la fecha, en forma individual y en reuniones en pequeños grupos, organizados según el área de trabajo y la especialidad. Los términos finales del documento fueron consensuados por todo el grupo de trabajo. Cada participante proporcionó su declaración de conflicto de intereses.
ConclusionesSe elaboró el Consenso Argentino para la Enfermedad de Pompe, considerando aspectos de la fisiopatología, la clínica, el diagnóstico y el tratamiento de esta enfermedad. Tratándose de una afección infrecuente, en la que los datos disponibles son limitados, las presentes recomendaciones deben ser consideradas como opinión de expertos.
Pompe disease (PD) is a rare autosomal recessive metabolic disorder which is caused by the absence or deficiency of the acid alpha-glucosidase lysosomal enzyme in the tissues of affected individuals.
ObjectiveThe objective of this consensus is to review the current guidelines and provide recommendations for a correct diagnosis, evaluation, management, and treatment of patients with PD.
MethodsWe organized a consensus with a foreign guest and national professionals experienced in PD in the areas of clinic, pediatric clinic, laboratory diagnosis, neuropathology, neumonology, nutrition, neurology, metabolic diseases, neuromuscular diseases (NMD) and rehabilitation of patients with MND. We conducted a literature review of the existing publications and articles relevant to EP up to date, individually and in small group meetings organized by field of work and specialty. The final terms of the document were agreed upon by the entire working group. Each participant provided their declaration of conflict of interests.
ConclusionsThe Argentine Consensus for Pompe disease was developed, considering aspects of the pathophysiology, clinical manifestations, diagnosis and treatment of this disease. Being a rare condition for which the available data are limited, these recommendations should be considered as expert opinion.
El presente Consenso Argentino para la Enfermedad de Pompe fue desarrollado con el auspicio de la Sociedad Neurológica Argentina y con la participación de miembros del grupo de trabajo en enfermedades del Sistema Nervioso Periférico. El mismo no pretende incluir todos los posibles procedimientos existentes, sino solo aquellos que se consideraron de mayor relevancia. Tratándose de una enfermedad poco frecuente, los datos disponibles sobre la misma son limitados, por lo que estas recomendaciones deben ser consideradas como una opinión de expertos.
Vale la pena destacar que el seguimiento de este consenso no garantiza un resultado médico exitoso, debiendo el médico tratante guiarse por su juicio y experiencia personal a la hora de escoger los procedimientos diagnósticos y terapéuticos más oportunos en cada paciente en particular.
IntroducciónLas enfermedades de depósito lisosomal (EDL) son afecciones poco frecuentes en las que ocurre una anormal acumulación de sustratos dentro de los lisosomas, lo que produce diferentes trastornos en la estructura y la función de los tejidos afectados1. Se estima que se presentan en conjunto en 1 de cada 7.000-8.000 recién nacidos vivos2,3, aunque es probable que estén subdiagnosticadas, ya que existe en general escaso conocimiento sobre las mismas.
La enfermedad de Pompe (EP), también conocida como glucogenosis tipo ii o deficiencia de maltasa ácida (OMIM # 232300), fue la primera EDL descrita. Se produce por un trastorno autosómico recesivo que lleva a la ausencia o marcada deficiencia de la enzima lisosomal alfa-glucosidasa ácida (AGA) (EC 3.2.1.20) en los individuos afectados4.
La AGA lisosomal cataliza la degradación del glucógeno a glucosa y, cuando la actividad de la enzima es deficiente, este se acumula en el lisosoma. Al romperse los lisosomas, el glucógeno se vuelca al citoplasma, afectando así a los elementos contráctiles de la célula e impidiendo el normal funcionamiento de las fibras del músculo esquelético, liso y cardíaco5. Este proceso conduce a hipotonía y debilidad muscular, por lo que la EP es también una miopatía metabólica dentro de las enfermedades neuromusculares (ENM)6,7.
La tasa de acumulación de glucógeno —y, por lo tanto, la gravedad de los síntomas de la enfermedad— depende principalmente de la actividad enzimática residual. La misma suele ser inferior al 1% de la actividad normal en los niños afectados menores de 1 año, hasta el 10% en la forma juvenil y menor del 40% en adultos. El fenotipo de cada paciente está determinado además por factores nutricionales, el tipo de fibra muscular afectada, la actividad física, la edad de inicio de la enfermedad y los modificadores genéticos aún no bien comprendidos4,6,8.
La EP es un desorden multisistémico, con un amplio espectro de fenotipos clínicos y grados variables de progresión, síntomas de inicio y niveles de afectación de los distintos órganos9. Dentro de esta variedad de presentaciones, se reconocen 2 extremos: una forma de inicio infantil y rápida progresión, y otra de inicio tardío y progresión más lenta. Se diferencian así 2 categorías muy amplias, la EP infantil y la EP de inicio tardío10. En ocasiones, sin embargo, los límites entre estas 2 formas clínicas no son tan evidentes.
Si bien la incidencia global de EP es variable dentro de las diferentes poblaciones estudiadas, la misma se estima en 1:40.0006,11. Los datos varían de 1:14.000 a 1:250.000 nacidos vivos, dependiendo del grupo étnico y geográfico12. Teniendo en cuenta la incidencia esperada, se estima que la EP es una entidad subdiagnosticada.
Historia de la enfermedad de PompeJohannes Cassianus Pompe (1901-1945) fue un patólogo holandés, formado en la Universidad de Utrecht y entrenado en Ámsterdam. En el año 1932, realizó la primera descripción conocida de la enfermedad que luego llevaría su nombre, en una niña de 7 meses de edad que falleció con severa debilidad muscular y una miocardiopatía hipertrófica. El análisis microscópico mostró depósito de glucógeno no solo en el corazón, sino también en el hígado, los riñones y el músculo esquelético. En 1936, Pompe obtuvo el título de Doctor en Medicina en la Universidad de Ámsterdam por su disertación sobre el tema «Cardiomegalia glucogénica». Durante la segunda guerra mundial, participó en forma heroica en la resistencia holandesa. Fue finalmente arrestado y más tarde ejecutado por las fuerzas de ocupación13-16.
En el año 1954, la enfermedad descrita por Pompe fue clasificada dentro de las enfermedades por trastorno del metabolismo del glucógeno como glucogenosis tipo ii17. En 1963, H.G. Hers demostró la ausencia de la enzima AGA en los lisosomas de los enfermos, lo que llevó a que la EP fuera reconocida como el primer desorden de almacenamiento lisosomal4,16. Entre 1960 y 1970, Engel et al. realizaron las primeras descripciones en pacientes de mayor edad (EP de comienzo tardío)18.
En 1979, se identificó el gen de la enzima AGA en el cromosoma 1719.
Los intentos iniciales de tratamiento específico para la EP con reemplazo enzimático obtenido de derivados placentarios se habían realizado en 1973. Estas pruebas se encontraron con dificultades por el desarrollo de respuesta inmunológica y por una limitada disponibilidad de la enzima20. En la década de 1990, la aparición de nueva tecnología permitió la producción de la suficiente cantidad de AGA recombinante (AGAr) como para que pudieran llevarse a cabo estudios clínicos de tratamiento con esta enzima. En el año 2006, la Food and Drug Administration de los Estados Unidos (FDA) y las Agencias Médicas Europeas aprobaron el uso de la AGA como el primer tratamiento específico para la EP. Esto fue basado en los resultados de un estudio inicial en 8 niños menores de un año, que mostró mejoría en la miocardiopatía, el crecimiento y las funciones motoras, y una prolongación en la sobrevida libre de ventilación asistida21.
Los resultados del primer estudio aleatorizado, controlado, doble ciego, de terapia de reemplazo enzimático (TRE) en EP de inicio tardío (Late onset treatment study [LOTS])22 llevaron a la aprobación en Estados Unidos de la TRE para el tratamiento de esta forma de la enfermedad. Junto a la aprobación, la FDA indicó control posmarketing de la enzima y la realización de estudios que se están llevando a cabo sobre aspectos inmunológicos, de seguridad, farmacocinéticos y de seguimiento a largo plazo acerca del papel de la AGA en el tratamiento de la EP23.
PatofisiologíaEl gen de la AGA se encuentra ubicado en el brazo largo del cromosoma 17 (17q25.2-q25.3)24. Se han identificado más de 350 mutaciones del mismo, que están registradas en el Pompe Center del Erasmus MC de Rotterdam (Pompe Center. Molecular aspects: mutations, http://cluster15.erasmusmc.nl/klgn/pompe/mutations.html [7 Aug 2013]). La mutación más frecuente en pacientes caucásicos es IVS1 (-32-13T-›G), hallada en alguno de los alelos en alrededor del 50% de los afectados.
La AGA lisosomal cataboliza la degradación del glucógeno contenido en la organela en glucosa y su deficiencia produce acumulación del mismo en múltiples tejidos. La rotura de la membrana lisosomal provoca la liberación del glucógeno acumulado, así como la extravasación del contenido del lisosoma al citosol. De modo que en la EP, la acumulación del glucógeno es tanto intra como extralisosomal25,26.
La naturaleza de la EP es compleja y va más allá de una simple enfermedad de almacenamiento del glucógeno27. Se han propuesto otros mecanismos de daño tisular, entre ellos un fracaso de la autofagia (proceso intracelular por el cual las macromoléculas y organelas son llevadas hacia los lisosomas para su degradación y reciclado)16. Esto provocaría una acumulación progresiva de los autofagosomas, que alteraría el aparato contráctil de las fibras musculares28. El exceso de autofagosomas se demostró tanto en pacientes con EP como en modelos de animales knockout para el gen de la AGA, y es en algunas fibras la única alteración encontrada16. También se ha indicado que los trastornos en la autofagia estarían relacionados con la existencia de alteraciones no bien caracterizadas en un subgrupo específico de lisosomas16.
Formas clínicasForma de comienzo infantilSe define como EP infantil a aquella cuyos síntomas se manifiestan antes del año de vida, independientemente de la severidad del cuadro. En estos casos, la actividad de AGA está ausente casi por completo, con menos del 1% de su valor normal en fibroblastos de la piel6,10.
El pronóstico fatal a corto plazo en ausencia de tratamiento con TRE impone la necesidad de su reconocimiento temprano. La edad media de inicio de los síntomas es de 4 meses, y la necesidad de asistencia respiratoria mecánica suele ocurrir alrededor de los 5,9 meses. Sin TRE, la muerte sobrevendría a los 8,7 meses29,30, en general, a causa de insuficiencia cardiorrespiratoria7,10,29. Puede decirse que existen 2 variantes. En su forma clásica, la EP infantil se presenta con miocardiopatía. Este cuadro, descrito inicialmente por Pompe14, corresponde al extremo más grave del espectro de la enfermedad. La edad media de inicio de los síntomas es 1,6 meses, la de diagnóstico varía entre 4,5 y 5,3 meses, y la de muerte entre 6,0 y 7,7 meses10,29. Es una enfermedad multisistémica y rápidamente progresiva, que causa compromiso muscular (esquelético, cardíaco y liso), severa hipotonía y dificultad en la deglución. Todas estas manifestaciones confluyen en compromiso respiratorio. Existe además afectación gastrointestinal y compromiso de las células del asta anterior. Puede haber macroglosia, hepatoesplenomegalia y retraso de crecimiento10,30. Las manifestaciones clínicas más frecuentes son marcada hipotonía con hipo-arreflexia, debilidad de músculos faciales, miocardiopatía severa con cardiomegalia, insuficiencia ventilatoria e infecciones respiratorias recurrentes. Los niños presentan retraso de crecimiento y de los hitos madurativos, así como dificultad en la alimentación y succión. La creatincinasa (CK) se halla habitualmente elevada hasta unas 10 veces del valor normal10,29,31-33.
Cuando no hay compromiso cardíaco, se habla de una forma atípica o variante muscular de la EP infantil27, en la que la progresión de la enfermedad es más lenta34.
Forma de comienzo tardíoEs aquella en que la sintomatología aparece luego del año de vida27. Este término puede ser confuso, ya que la EP de inicio tardío incluye a pacientes cuya sintomatología comienza tanto en la niñez como en la juventud y la adultez, aun la avanzada27. Esta categoría constituye, por lo tanto, un grupo heterogéneo de individuos con afección preponderante del músculo esquelético y poca evidencia de lesión cardíaca6, aunque puede haber anormalidades electrocardiográficas y arritmias12.
En la EP de inicio tardío, la actividad enzimática se conserva parcialmente en fibroblastos de la piel, hasta un 40% de los valores normales6,10.
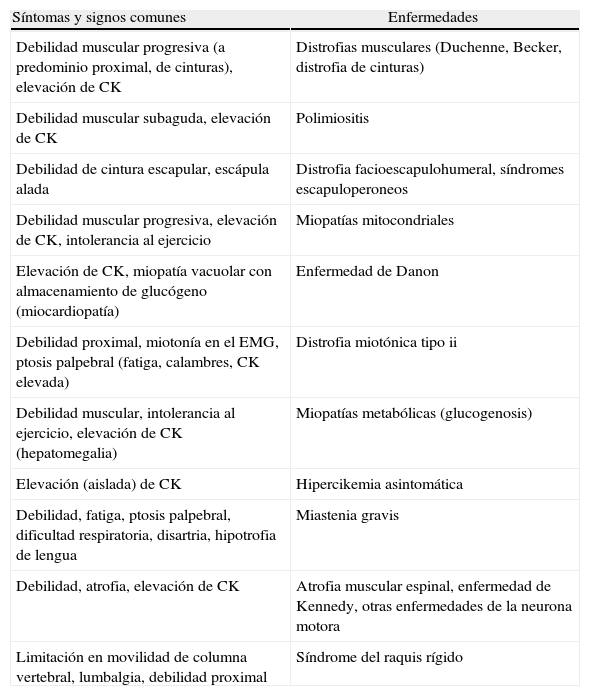
Este cuadro comparte características clínicas con muchas otras ENM con las que puede ser confundido, como las distrofias musculares, en especial las de las cinturas y la facioescapulohumeral, la polimiositis, las miopatías metabólicas, la miastenia gravis y las enfermedades de neurona motora35, y también debe considerarse en caso de hiperckemia asintomática36,37 (véase la tabla 1). Sin embargo, el patrón de afectación muscular y su distribución, asociados a la secuencia de aparición de la debilidad en los distintos grupos musculares, le confiere una característica casi única. Por este motivo, su diagnóstico requiere un alto índice de sospecha clínica y para ello es preciso un cuidadoso interrogatorio y examen neuromuscular. Se han reportado demoras en el diagnóstico de hasta 30 años38.
Principales diagnósticos diferenciales de la enfermedad de Pompe
| Síntomas y signos comunes | Enfermedades |
| Debilidad muscular progresiva (a predominio proximal, de cinturas), elevación de CK | Distrofias musculares (Duchenne, Becker, distrofia de cinturas) |
| Debilidad muscular subaguda, elevación de CK | Polimiositis |
| Debilidad de cintura escapular, escápula alada | Distrofia facioescapulohumeral, síndromes escapuloperoneos |
| Debilidad muscular progresiva, elevación de CK, intolerancia al ejercicio | Miopatías mitocondriales |
| Elevación de CK, miopatía vacuolar con almacenamiento de glucógeno (miocardiopatía) | Enfermedad de Danon |
| Debilidad proximal, miotonía en el EMG, ptosis palpebral (fatiga, calambres, CK elevada) | Distrofia miotónica tipo ii |
| Debilidad muscular, intolerancia al ejercicio, elevación de CK (hepatomegalia) | Miopatías metabólicas (glucogenosis) |
| Elevación (aislada) de CK | Hipercikemia asintomática |
| Debilidad, fatiga, ptosis palpebral, dificultad respiratoria, disartria, hipotrofia de lengua | Miastenia gravis |
| Debilidad, atrofia, elevación de CK | Atrofia muscular espinal, enfermedad de Kennedy, otras enfermedades de la neurona motora |
| Limitación en movilidad de columna vertebral, lumbalgia, debilidad proximal | Síndrome del raquis rígido |
CK: creatincinasa; EMG: electromiograma.
La enfermedad se caracteriza por debilidad muscular lentamente progresiva de predominio proximal. La debilidad de los miembros inferiores es el síntoma de presentación más frecuente en la EP del adulto, representando más del 90% de los casos. La fuerza muscular declina con más rapidez en los miembros inferiores que en los superiores39. Las primeras manifestaciones suelen pasar inadvertidas y afectan a los músculos axiales, abdominales y paravertebrales, que se encuentran comprometidos en forma precoz35. En los miembros inferiores, los músculos del compartimento posterior del muslo35 y los glúteos están característicamente afectados, mientras que los cuádriceps preservan la fuerza por más tiempo9. Los músculos distales se conservan hasta un estadio bastante más avanzado. En los miembros superiores, la atrofia de músculos periescapulares es importante en algunos casos y pueden observarse escápulas aladas. El bíceps braquial y los músculos distales están, en general, más preservados hasta estadios avanzados.
Los flexores del cuello están generalmente comprometidos9. Esto puede asociarse a escoliosis y espina rígida40-42. En ocasiones, los pacientes refieren mialgias y dolor lumbar38. Estos síntomas, así como la fatiga fácil y la intolerancia al ejercicio, son comunes en estadios iniciales o preclínicos35,38.
La debilidad en la lengua está siempre presente, aunque solo en ocasiones provoca disartria43. La ptosis palpebral uni o bilateral9,44,45 y la debilidad facial46 son manifestaciones menos frecuentes. La disfagia puede aparecer en estadios avanzados10,44. En los niños en la primera infancia se observan retraso en el desarrollo motor y trastornos en el crecimiento, con dificultad en la ganancia de peso en los adolescentes.
Es característico el compromiso precoz del diafragma y los músculos respiratorios accesorios, lo que lleva a una falla respiratoria relativamente temprana, aun en pacientes que se encuentran ambulatorios33,35, e incluso antes de la aparición de debilidad significativa en cualquier otro grupo muscular47 La insuficiencia respiratoria es la principal causa de morbimortalidad en esta enfermedad48 y es la manifestación inicial de la misma en hasta un tercio de los pacientes35. La edad de la muerte varía desde la primera infancia hasta la adultez avanzada, en función de la tasa de progresión de la enfermedad, el grado de compromiso de los músculos respiratorios y la presencia de otras comorbilidades6,49. La variabilidad de la presentación clínica en este grupo es muy marcada, incluyendo individuos asintomáticos50.
Compromiso respiratorio en la enfermedad de PompeLa forma infantil de la EP tiene un compromiso respiratorio más rápidamente progresivo que la forma de comienzo tardío7,39. Pero en la primera, la miocardiopatía suele marcar la evolución clínica, mientras que en la forma de comienzo tardío el corazón no suele estar afectado31,51. Los síntomas respiratorios pueden ser el hallazgo inicial en 30% de los casos con EP de inicio tardío, e incluyen ortopnea, disnea de ejercicio, síntomas derivados de las alteraciones respiratorias durante el sueño33,52 y de las infecciones respiratorias debidas a tos débil.
Las manifestaciones motoras y respiratorias no progresan del mismo modo. Un paciente ambulatorio puede necesitar ventilación nocturna, mientras que otro en silla de ruedas puede tener función pulmonar normal. Esta es una de las características de la EP. La debilidad diafragmática, las alteraciones respiratorias durante el sueño y la insuficiencia respiratoria son también habituales en el curso de la EP. Esta última puede ser de comienzo agudo o insidioso. El compromiso de los músculos respiratorios es la causa más común de muerte prematura en pacientes con EP53.
Evaluación clínica respiratoriaLas manifestaciones respiratorias son consecuencia de la debilidad de los músculos inspiratorios (en particular, el diafragma), de la debilidad de los músculos espiratorios, de las alteraciones respiratorias durante el sueño33,52 y de las alteraciones del control respiratorio que se ponen de manifiesto durante ese período54. Aproximadamente, la mitad de los pacientes adultos reportan disnea de esfuerzo al inicio de la enfermedad55.
La ortopnea es altamente indicativa de debilidad diafragmática. La presencia de respiración paradojal del abdomen, asociada con ortopnea, es muy indicativa de paresia o parálisis diafragmática, un hallazgo a veces precoz en la EP, que puede ser, en ocasiones, la primera manifestación de la enfermedad24,56-59. La incapacidad para alimentarse en las formas infantiles debe ser vista como un equivalente de la disnea de esfuerzo de los adultos.
Frecuentemente, las manifestaciones más precoces del compromiso del sistema respiratorio son la debilidad de los músculos espiratorios y la tos alterada, las cuales son responsables de los repetidos episodios de traqueobronquitis y neumonía. La presencia de cefaleas matutinas, hipersomnia diurna, ronquidos, jadeo (gasping) o sueño inquieto (restless sleep) son indicativas de trastornos respiratorios del sueño (TRS)56,60-62. En términos generales, en las ENM progresivas los TRS aparecen asociados a una capacidad vital (CV) del 30-50% del predicho; sin embargo, en la EP, estos TRS pueden ocurrir cuando la CV sentado está sólo moderadamente disminuida, debido al desproporcionado compromiso diafragmático10,23,63-65.
Evaluación funcional respiratoriaLa evaluación de la función respiratoria debe ser efectuada en forma periódica10,25,59, independientemente del grado de debilidad muscular periférica66. Se recomienda al menos una evaluación respiratoria anual en la EP tardía, sin importar el grado de compromiso de la función muscular periférica67. La evaluación respiratoria debe estar dirigida a la valoración del trastorno restrictivo y de la función del diafragma, la detección de tos débil, la presencia de hipoventilación alveolar y de TRS.
En los pacientes con EP de inicio tardío, la evaluación de la función pulmonar debe incluir la medición espirométrica de la CV, el volumen espiratorio forzado en un segundo, la presión inspiratoria máxima (PImáx) y la presión espiratoria máxima (PEmáx)62,68. Estas últimas son más sensibles que la CV para detectar debilidad y permiten discriminar la presencia de debilidad inspiratoria y espiratoria por separado69. La CV en decúbito dorsal es una manera simple de evaluar la función del diafragma y debería ser sistemáticamente efectuada en la EP de inicio tardío67. La medición de flujo espiratorio pico permite la evaluación objetiva de la fuerza muscular espiratoria en ausencia de obstrucción bronquial. La estimación de la saturación arterial de O2 mediante pulsioximetría y del CO2 arterial mediante la capnografía completa la evaluación y puede limitar la utilización de la punción arterial.
La CV y la disfunción diafragmática son 2 importantes predictores de la insuficiencia respiratoria y de la hipoventilación crónica durante el sueño24,56,70. El compromiso del diafragma puede ser un hallazgo precoz en la EP, pudiendo algunas veces ser la primera manifestación clínica de la enfermedad24,55-59. Antes de que se produzca una reducción de la CV en posición sentada, se puede evidenciar la debilidad diafragmática midiendo la CV en decúbito dorsal67,70. Una disminución del 10% de la CV en decúbito dorsal ya indica debilidad diafragmática.
En el caso particular de los lactantes, se torna muy compleja la valoración objetiva de la función pulmonar, por lo que la clínica toma relevancia, debiéndose evaluar el grado de fatigabilidad (evaluar la capacidad para alimentarse sin aumentar el trabajo respiratorio). La espirometría no es una opción y las pruebas de función pulmonar pueden requerir sedación. La CV del niño puede estimarse durante el llanto y la medición del flujo inspiratorio negativo podría aproximar una medida de la fuerza muscular inspiratoria71. Particularmente en la EP de inicio infantil son muy comunes las atelectasias del lóbulo inferior izquierdo, debido a la compresión que ejerce el corazón hipertrofiado sobre el bronquio fuente izquierdo.
Trastornos respiratorios del sueñoDurante el sueño, pueden ocurrir diversos trastornos en la respiración54,56,61,62. Los TRS que se presentan a menudo en los pacientes con EP son la hipoventilación sostenida y la apnea obstructiva del sueño23,56,61-63,65. Los TRS en la EP pueden ocurrir aun cuando la CV en posición sentado esté solo moderadamente disminuida56,70. Los pacientes pueden llegar a la hipercapnia y la hipoxemia23,63,64,72-74. La desaturación y la hipoventilación durante el sueño56 pueden requerir soporte ventilatorio39.
En la EP infantil, los TRS pueden desarrollarse antes de que los padres noten los síntomas75. Todos los pacientes con EP deben tener una historia detallada de las características del sueño en el momento del diagnóstico y durante el seguimiento23,76.
Insuficiencia respiratoriaA medida que avanza la enfermedad, se van manifestando —primero durante el sueño y luego durante la vigilia— la hipoxemia (disminución de la PO2 arterial y de la saturación de oxígeno) y la retención crónica de CO2 (PCO2 arterial elevada y aumento del bicarbonato sérico). Como la participación diafragmática es un hallazgo temprano, la insuficiencia respiratoria puede desarrollarse aún en pacientes ambulatorios56-60,77-80. En los pacientes con EP de inicio tardío, la insuficiencia respiratoria solo es percibida mínimamente cuando la debilidad muscular limita la capacidad de ejercicio. La insuficiencia respiratoria es la causa más común de muerte en pacientes con EP de aparición tardía55.
Para valorar el desarrollo de insuficiencia respiratoria, se recomienda estimar de rutina el intercambio de gases realizando una oximetría de pulso y una capnografía. Si son normales, no resultaría necesario evaluar gases en sangre arterial. Si no se pudiera realizar la capnografía, se debería obtener un valor de PCO2 (o bicarbonato) en sangre venosa y/o realizar un dosaje de PCO2 en sangre capilar, para evaluar la hipoventilación alveolar10,81.
Las radiografías de tórax se deben obtener en el momento del diagnóstico y cada vez que haya algún deterioro clínico. También son útiles para determinar la presencia de atelectasias y dirigir la asistencia kinésica respiratoria.
TratamientoEl tratamiento de las anomalías respiratorias presentes en los pacientes con EP y otras ENM requiere de un neumonólogo con experiencia en estas enfermedades, así como de un equipo multidisciplinario compuesto esencialmente por kinesiólogos respiratorios82.
Vacunación: además de las vacunas regulares, todos los pacientes deben recibir las vacunas correspondientes a neumococo e influenza10.
Manejo de las secreciones: para facilitar el aclaramiento de las mismas, se deben utilizar técnicas manuales y/o mecánicas de limpieza y de tos asistida75,83,84.
Fármacos: el tratamiento de las infecciones pulmonares debe ser precoz y agresivo10,83.
No debe ser el oxígeno el tratamiento de la hipoventilación alveolar pura, es decir, sin anomalías del intercambio gaseoso asociadas. De manera que el tratamiento debe dirigirse al trastorno respiratorio subyacente. Para los pacientes con apnea obstructiva del sueño, el tratamiento puede limitarse a la presión positiva continua; en pacientes con hipoventilación nocturna, el tratamiento es ventilación no invasiva (BiPAP)56,65. La modalidad de BiPAP se debe considerar si la PCO2 es > 45mmHg, si la CV en posición supina es < 50% de lo previsto, si la fuerza inspiratoria negativa es <60cm de H2O, o si la saturación de oxígeno es <88% (por 5 min seguidos) durante el sueño85,86.
Si estos métodos no lograran corregir la hipoxia, se puede utilizar oxígeno suplementario, cuidando su tasa de administración junto con el nivel arterial de dióxido de carbono, para evitar la hipercapnia inducida por oxígeno87.
Un trabajo reciente demostró que la utilización de la enzima recombinante puede permitir la estabilización de la función respiratoria y motora22. Estudios de seguimiento demostraron que este efecto se mantuvo en el tiempo88. Otros estudios sobre los efectos de la TRE a largo plazo reportan similares beneficios89, lo que ha sido confirmado mediante revisión sistemática90.
La tabla 2 resume las principales recomendaciones para el tratamiento de los desórdenes respiratorios en la EP de inicio tardío.
Recomendaciones para el manejo y el tratamiento de los desórdenes respiratorios en la enfermedad de Pompe de comienzo tardío
| El seguimiento debe realizarse preferentemente por un neumonólogo con experiencia en ENM y un equipo multidisciplinario que incluya kinesiólogos respiratorios |
| La evaluación de la función respiratoria debe efectuarse en forma periódica, al menos una vez al año, independientemente del grado de debilidad muscular periférica |
| Debe incluir la medición espirométrica de la CV, el VEF1, la PImáx, la PEmáx y la CV en decúbito dorsal para evaluar la función del diafragma |
| Se recomienda realizar de rutina una oximetría de pulso y una capnografía. Si la capnografía no está disponible, obtener un valor de PCO2 (o bicarbonato) en sangre venosa y/o PCO2 en sangre capilar, para evaluar hipoventilación alveolar |
| Las radiografías de tórax se deben obtener al momento del diagnóstico y cuando exista algún deterioro clínico |
| Mantener al día el esquema de vacunación, incluyendo la vacuna contra neumococo e influenza |
| El tratamiento de las infecciones pulmonares debe ser precoz y agresivo |
| Mantener la vía aérea libre de secreciones utilizando técnicas manuales y/o mecánicas de limpieza y de tos asistida |
| Tratar los desórdenes respiratorios relacionados con el sueño con equipos de la CPAP o con BiPAP en caso de hipoventilación nocturna |
| La modalidad de BiPAP se debe considerar si la PCO2 es mayor a 45mmHg, la CV en posición supina es <50% de lo previsto, la fuerza inspiratoria negativa es <60cm de H2O o la saturación de oxígeno es <88% (por 5 min continuos) durante el sueño. Si estos métodos no corrigen la hipoxia, se puede utilizar oxígeno suplementario, cuidando su tasa de administración junto con el nivel arterial de dióxido de carbono para evitar la hipercapnia |
| La utilización de TRE puede permitir la estabilización de la función respiratoria y motora, con un efecto sostenido en el tiempo |
BiPAP: ventilación no invasiva; CV: capacidad vital; CPAP: presión positiva continua; ENM: enfermedades neuromusculares; PEmáx: presión espiratoria máxima; PImáx: presión inspiratoria máxima; TRE: terapia de reemplazo enzimático; VEF1: volumen espiratorio forzado en un segundo.
La forma infantil clásica de la EP se presenta en general con cardiomiopatía hipertrófica asociada a hipotonía y falla respiratoria10. La hipertrofia es secundaria a la acumulación de glucógeno en el miocardio y puede llegar a obstruir el tracto de salida del ventrículo izquierdo33,91. El compromiso cardíaco puede fomentar la insuficiencia respiratoria, tanto por la alteración hemodinámica como por el tamaño cardíaco, que afecta a la capacidad pulmonar.
Con la enfermedad librada a su propio curso, la cardiopatía progresa en forma casi invariable a miocardiopatía dilatada. La resonancia magnética (RM) cardíaca ha podido identificar y cuantificar la hipertrofia biventricular midiendo parámetros hemodinámicos en pacientes con EP infantil. Por medio de la utilización de contraste, se han podido identificar también áreas con fibrosis miocárdica92. Es posible observar arritmias atribuibles al efecto aislante del glucógeno en el tejido de conducción y a isquemia subendocárdica, debido a la masiva hipertrofia y la insuficiencia coronaria relativa que se establece74. La combinación de arritmias con la cardiomiopatía hipertrófica puede determinar muerte súbita. La TRE se ha mostrado capaz de revertir la enfermedad, mejorando la función y reduciendo los diámetros cardíacos de los pacientes93.
El tratamiento de los trastornos cardiológicos incluye el uso de fármacos como diuréticos, inhibidores de la enzima conversora de angiotensina y digoxina, entre otros. Estas deben ajustarse a cada caso y se recomienda que el manejo terapéutico sea encarado por cardiólogos experimentados.
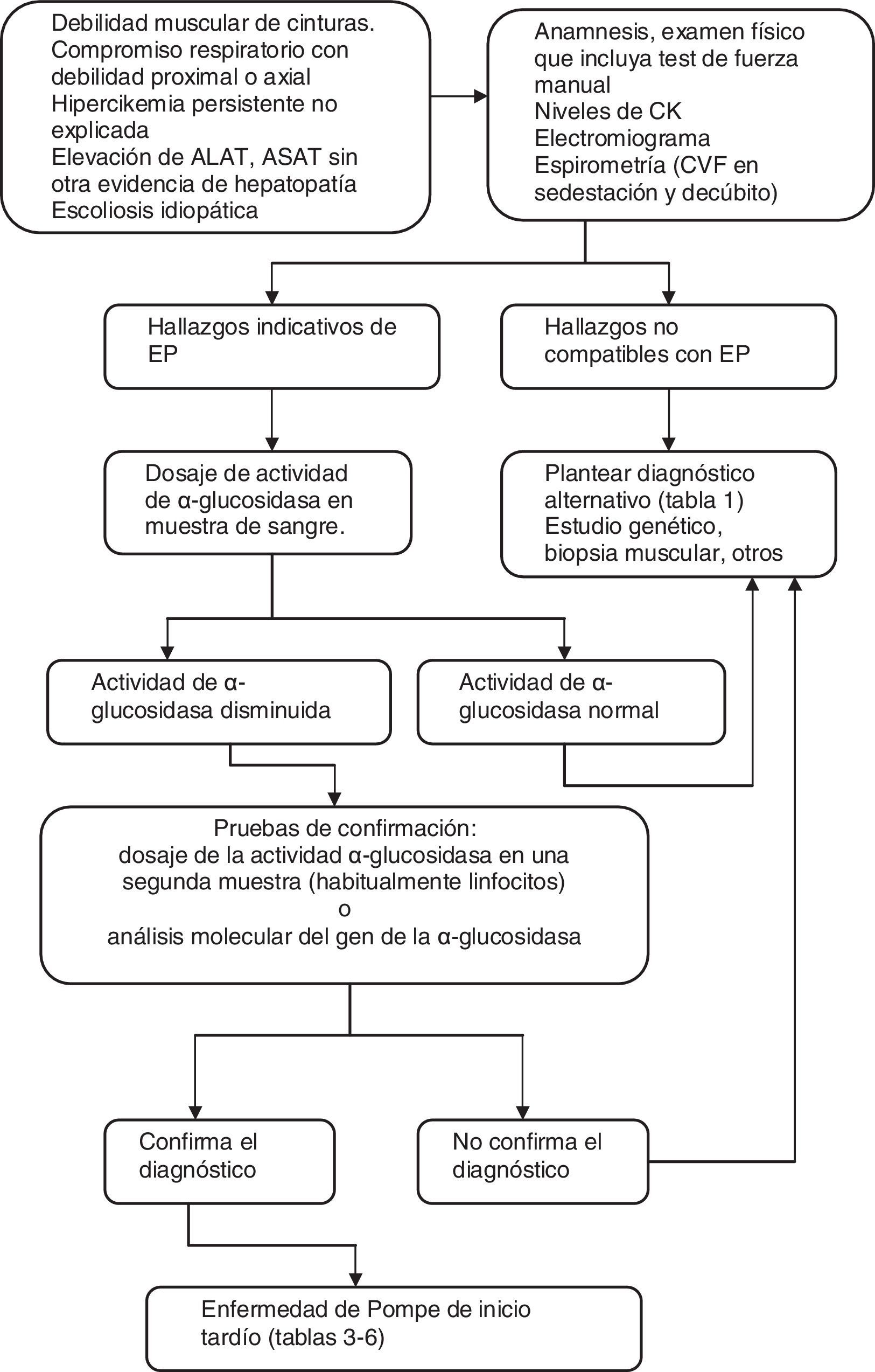
Métodos complementarios de diagnósticoAnte la sospecha clínica, pueden utilizarse distintas pruebas de laboratorio que permitan demostrar la disminución de la actividad enzimática de la AGA o la mutación del gen responsable que pueda originarla.
La investigación de la actividad enzimática en gota seca es, por su accesibilidad y disponibilidad, el primer test diagnóstico al que se debe recurrir. El resultado debe ser confirmado en un segundo ensayo sobre otro tejido: linfocitos, leucocitos, músculo. El diagnóstico sobre fibroblastos brinda información esencial sobre el pronóstico de la enfermedad.
Ante un resultado no concluyente o negativo, es necesario efectuar una biopsia muscular. Esta podrá aportar elementos que apoyen la hipótesis diagnóstica o será útil para establecer un diagnóstico alternativo25.
Diagnóstico de sujetos en riesgoTeniendo en cuenta el carácter recesivo de la enfermedad, y la posibilidad de que la misma se mantenga silente por mucho tiempo, es importante que los familiares en riesgo de los pacientes con diagnóstico confirmado, en especial los hermanos, sean estudiados para descartar su afectación y decidir luego de la evaluación clínica la necesidad de TRE.
Análisis de la actividad enzimáticaLa actividad de la AGA medida en cultivo de fibroblastos obtenidos de una biopsia de piel es considerada el gold standard para el diagnóstico de EP en pacientes sintomáticos. Esta metodología brinda información valiosa en cuanto a la actividad enzimática residual, permitiendo discriminar entre las formas severas y las moderadas de la enfermedad. Por ser un método lento (el diagnóstico puede demorar hasta 6 semanas) e invasivo, se han desarrollado otros ensayos para medir la actividad de AGA en sangre94.
La actividad de la AGA también se puede medir en linfocitos purificados, músculo, leucocitos o gotas de sangre seca sobre papel de filtro (GSPF), pero en estos tejidos se corre el riesgo de que la isoenzima maltasa-glucoamilasa interfiera en la medición de la actividad enzimática tomada con pH ácido6. El uso de acarbosa, que inhibe la isoenzima maltasa-glucosamilasa, ha permitido aumentar la selectividad del método y obtener así resultados confiables27,29,30. La detección de la actividad enzimática se puede realizar por métodos fluorométricos o por espectrometría de masa en tándem94. Las muestras son susceptibles a condiciones ambientales extremas durante el envío, la conservación y el transporte, por lo que si no se toman las debidas precauciones la actividad enzimática se verá alterada, dando la posibilidad de resultados falsos positivos o no concluyentes.
Las GSPF pueden ser obtenidas fácilmente y enviadas desde lugares remotos hasta centros de referencia para su posterior análisis, siendo especialmente útiles para la detección rápida en neonatos y niños94. La recolección de la muestra es un paso importante en la seguridad y eficiencia de la determinación. Un video de la técnica de recolección de GSPF está disponible en línea (Fundación para el Estudio de las Enfermedades Neurometabólicas, 2011). La fecha de la toma siempre debe tenerse en cuenta en la tarjeta para ayudar en la interpretación de los resultados. Las tarjetas deben ser secadas en posición horizontal a temperatura ambiente durante al menos 4 h y almacenadas a 4°C después del secado.
El método de espectrometría de masa en tándem fue desarrollado para su uso en programas de cribado o tamizado neonatal, y tiene el potencial para ser incorporado a futuro en un panel de estudios de enfermedades lisosomales95.
Estudios bioquímicos complementarios a la actividad enzimáticaExisten 2 tetrasacáridos, Hex4 y Glc4, cuyas concentraciones en plasma y orina se encuentran aumentadas en pacientes con EP. No son biomarcadores específicos de esta enfermedad, ya que también se encuentran elevados en otras glucogenosis94. Se ha demostrado el descenso de la concentración de estos indicadores en pacientes bajo TRE96.
Biopsia muscularLa forma clásica infantil presenta una severa miopatía vacuolar con acumulación de glucógeno, evidente con técnica de periodic acid schiff (PAS). Existe disminución de miofibrillas, observable con miosina ATPasa. Las vacuolas son positivas con fosfatasa ácida (marcador lisosomal y de autofagia). Ambos tipos de fibras pueden estar comprometidas. A nivel ultraestructural, se suele observar glucógeno intra y extralisosomal, distorsión de la estructura con disminución notable de miofibrillas y vacuolas autofágicas.
En casos de EP de comienzo tardío, los hallazgos pueden presentar un rango amplio de variación. Las biopsias pueden ser normales, tener cambios mínimos detectables, como una vacuola lineada o no lineada fosfatasa ácida positiva, o bien características severas similares a las descritas en la forma infantil. Puede observarse aislada fibra necrótica y ha sido reportado un patrón neurogénico en algunos casos45,97-99. Los hallazgos también dependen del músculo biopsiado, lo que determina la variabilidad de las observaciones. Es esencial evaluar la biopsia de un músculo comprometido clínicamente.
Una biopsia normal o inespecífica no excluye EP6,97,98 y una biopsia con hallazgos mínimos, como una sola vacuola lineada o no lineada fosfatasa ácida positiva, debe ser estudiada con microscopia electrónica y chequeada la AGA en papel de filtro antes de proseguir con la investigación de otro tipo de miopatías. La positividad de la biopsia no solo depende del músculo biopsiado, sino también del momento evolutivo de la enfermedad97,98. La biopsia muscular puede ser de importancia decisiva en aquellos casos en donde los valores de las otras determinaciones, como el estudio en gota de sangre seca o los dosajes, arrojen resultados dudosos o ambiguos en el contexto de un cuadro clínico compatible, tanto para su evaluación morfológica como para el dosaje de AGA sobre el músculo.
Estudios moleculares (ADN)El gen AGA contiene 20 exones y solo el primero no es codificante. Se localiza en el cromosoma 17q25100. El ADN codificante (ADNc) codifica un polipéptido de 952 aminoácidos, incluyendo un péptido señal de 27 aminoácidos. Más de 453 alteraciones en el gen AGA se han descrito hasta ahora en el Centro de Pompe (www.pompecenter.nl), entre ellas 135 mutaciones patogénicas, 97 mutaciones no patogénicas y 46 variantes de la secuencia con efecto funcional desconocido. Las alteraciones incluyen mutaciones sin sentido, de sitio de corte y empalme, y reorganizaciones parciales de genes, incluso pérdidas intragénicas pequeñas, grandes e inserciones.
Existen mutaciones muy frecuentes, como la de corte y empalme (splicing) IVS1 (-32-13T-›G), que da cuenta de casi el 50% de las alteraciones halladas en la EP de inicio tardío en los niños caucásicos y adultos con EP10.
Las correlaciones genotipo-fenotipo no se hallan bien definidas, aunque la homocigosidad para mutaciones que truncan está casi siempre asociada con EP. Otros factores (genéticos y ambientales) pueden influir en el fenotipo, lo que indica que pacientes con las mismas mutaciones en el gen AGA pueden presentar fenotipos clínicamente diferentes101.
El análisis de las mutaciones del gen AGA es relevante para el diagnóstico prenatal, siempre que el genotipo del paciente índice sea conocido, y en la identificación de individuos portadores, cuando una mutación familiar es conocida (si se sabe cuáles son las mutaciones familiares, el examen molecular es el estudio ideal). También lo es para confirmar el diagnóstico en pacientes heterocigotas con EP de aparición tardía.
El análisis de las mutaciones del gen AGA es relevante además para consejo genético. Siendo una enfermedad autosómica recesiva, los padres de un individuo afectado son portadores y presentan un riesgo de recurrencia del 25%. De esto se desprende que se debe ofrecer asesoramiento genético a todos los padres de un niño con EP, así como a todos los adultos con EP, obteniendo un árbol genealógico de 3 generaciones como mínimo.
Para la identificación de miembros de la familia que puedan ser portadores, se requiere también un análisis mutacional del ADN.
Otros exámenes complementariosEnzimas musculares: la CK puede estar elevada, pero no suele sobrepasar valores de 1.500 U o 2.000 U102. Las transaminasas hepáticas pueden estar elevadas en forma persistente, aún con niveles de CK solo discretamente superiores a los normales. La elevación conjunta de estas enzimas se considera muy sensible para detectar el daño muscular en la EP103.
Electromiograma: en las formas infantiles, muestra irritabilidad de membrana y actividad denervatoria, siendo características las descargas repetitivas de alta frecuencia, que se consideran secundarias al compromiso del asta anterior. Pueden encontrarse, en ausencia de miotonía clínica, verdaderas descargas miotónicas. La contracción voluntaria muestra potenciales de unidad motora polifásicos breves y de baja amplitud104.
La mayoría de los pacientes con EP de inicio tardío tienen hallazgos compatibles con miopatía en músculos proximales sin presencia de actividad denervatoria104, o la misma se encuentra restringida a grupos específicos, en especial paravertebrales46. Estudios recientes indican que debería realizarse una evaluación de rutina de los músculos paravertebrales lumbares ante la sospecha diagnóstica de EP105.
Imágenes: se han mostrado confiables para determinar el patrón y la extensión del compromiso muscular. La RM se considera hoy en día el método de elección105,106. Como en la mayoría de las miopatías, las imágenes en T1 son suficientes para una adecuada evaluación diagnóstica106.
Los estudios muestran que la enfermedad tiende a progresar a lo largo de años desde el tronco a las extremidades, con mayor afectación de músculos axiales y del muslo que de la cintura escapular y las piernas105,106. El compromiso de los paravertebrales lumbares y el psoas, en particular, es universal, y suele haber afectación precoz de los músculos de la cintura abdominal. Los cambios miopáticos en estas regiones pueden observarse aún en individuos asintomáticos105.
Entre los músculos del tronco suelen estar más preservados los intercostales. La región facial se halla, en general, respetada, aunque la RM muestra severo compromiso de la lengua, en forma independiente de la severidad de la enfermedad. También hay compromiso precoz de los extensores del cuello106.
Es importante destacar que aunque los estudios por imágenes pueden indicar el diagnóstico de EP y ayudan a diferenciar el cuadro de otras miopatías, no reemplazan el valor de un adecuado interrogatorio y examen neurológico.
En la figura 1 se resumen las etapas habituales en el diagnóstico de la EP de inicio tardío.
Tratamiento
El tratamiento de los pacientes con EP incluye, por un lado, el manejo de los síntomas y las complicaciones derivados de la enfermedad y, por el otro, la implementación de una terapéutica específica que procura reponer la enzima faltante o deficitaria (TRE).
El seguimiento de estos pacientes debe involucrar, como en casi todas las ENM, distintas especialidades para diferentes problemas clínicos. El equipo de trabajo debe estar formado por médicos (neurólogo, clínico, neumonólogo, cardiólogo, nutricionista, gastroenterólogo), kinesiólogos y terapeutas físicos, ocupacionales y del lenguaje. Un enfoque multidisciplinario, con la coordinación de un neurólogo, es el más adecuado en estos casos.
Manejo de los trastornos musculoesqueléticosLos objetivos deben centrarse en la prevención de las complicaciones primarias y secundarias de la enfermedad. De modo similar que para otras ENM, se busca obtener la máxima función motora posible. Se procurará mejorar la marcha y las transferencias, subir y bajar escaleras, agacharse y levantarse, conservar la función respiratoria y mantener en todo lo posible la independencia para las actividades de la vida diaria. El objetivo final de la rehabilitación es, en última instancia, mejorar la calidad de vida del paciente.
Todos los tratamientos físicos deben estar adecuados a cada paciente y en relación con su estado en particular. Los mismos deberán centrarse en la situación actual y teniendo en cuenta las expectativas futuras10. La inclusión de complementos terapéuticos, tales como ejercicios rehabilitatorios de la deglución, foniátricos o terapia ocupacional, deben ser individuales en cada caso según se considere conveniente. La rehabilitación debe articularse con otras medidas de soporte, como las respiratorias, nutricionales y psicológicas, y el eventual uso de aparatos, ortesis o medios de desplazamiento.
Los tratamientos físicos en la EP son complementarios y necesarios en forma conjunta con la TRE cuando esta está indicada.
En las ENM en general, el uso de ejercicios contra resistencia ha sido tradicionalmente contraindicado ante la posibilidad de que ese tipo de práctica produzca un mayor deterioro en los músculos ya comprometidos, acelerando los mecanismos de degeneración107-109. Si bien en la EP hay pocos trabajos sobre el efecto de los ejercicios de fortalecimiento, algunos estudios indican el uso de ejercicios aeróbicos submáximos110,111.
Se ha reportado que la combinación de ejercicios de resistencia y aeróbicos resulta en la mejoría de la fuerza muscular y en el test de marcha de los 6 min en pacientes con EP de inicio tardío112. También se ha postulado como favorable el ejercicio aeróbico en otras varias ENM113-115.
La ubicación de los lisosomas entre el aparato contráctil de las fibras musculares podría convertir esas organelas en más vulnerables al daño producido por las contracciones116. Todos los pacientes con ENM deben ser informados de que el ejercicio intenso conlleva el riesgo de mayor daño y disminución de la función, e instruidos para poder advertir los signos de debilitamiento o fatiga por exceso de ejercicio. La aparición de calambres, sensación de pesadez o falta de aire debe ser tenida en cuenta. Hay que extremar los recaudos para evitar la fatiga muscular y los ejercicios que induzcan contracciones excéntricas de las fibras musculares o se realicen en condiciones anaerobias. Es importante enseñar al paciente a monitorear su frecuencia cardíaca y respiratoria en relación con el ejercicio y brindarle las pautas de alarma necesarias para su manejo. También las estrategias para conservar energía (como programar las actividades del día a día) y obtener ventajas biomecánicas (p. ej., el manejo de la velocidad de la marcha, la adecuación del hogar o la elevación de los asientos). El programa de ejercicios debe incluir la caminata, cinta, bicicleta, hidroterapia y natación. Hay que hacer énfasis en usar todos los grupos musculares, agonistas y antagonistas. Cuando se realicen ejercicios que involucran algún esfuerzo, deben tenerse las mismas precauciones que se recomiendan para otras ENM de tipo degenerativo117,118. No existe acuerdo acerca del mecanismo por el cual los ejercicios de resistencia serían beneficiosos en la EP. Algunos postulan que el ejercicio muscular continuo disminuiría el acúmulo de glucógeno lisosomal, mejorando así el rendimiento de esos músculos17, mientras que otros aseguran que esa mejoría no sería a través de la reducción del sustrato119. Los pacientes con EP menos comprometidos se benefician más que los más afectados con los tratamientos de terapia física120.
Se ha recomendado que los ejercicios empiecen lentamente, permitiendo períodos de descanso, seguidos por un aumento gradual en la intensidad de leve a moderada, hasta llegar a niveles aeróbicos de un 0 a un 70% del esfuerzo máximo, con una frecuencia de 3 a 5 días a la semana10,108,121. Es recomendable siempre el control neumonológico de los pacientes durante los programas de rehabilitación, ya que, como se mencionó, estos pacientes presentan particular compromiso respiratorio aún en etapas tempranas.
La realización de ejercicios durante la infusión de la TRE no demostró aún adicionar alguna ventaja para los pacientes122.
Las técnicas para tratar y prevenir contracturas y deformaciones articulares son las mismas que las utilizadas para otras ENM. Estas incluyen, por ejemplo, las elongaciones, la utilización de férulas o valvas y evitar posiciones inapropiadas123.
Debido a la afectación de los músculos espinales lumbares y a la tendencia a adoptar posturas compensatorias, se recomienda realizar ejercicios posturales preventivos y enseñar las mejores posturas en la vida diaria con un fin ergonómico. Además de la terapia física y el equipamiento ortésico, se debe recurrir a todas las medidas de soporte técnicas, adaptaciones del hogar y elementos para sustentar la movilidad, tales como bastones, andadores, soportes para la columna o el cuello, sillas de ruedas o scooters. Estas medidas deben ser evaluadas caso por caso por parte del equipo interviniente en el momento adecuado.
La tabla 3 resume los aspectos más importantes del manejo musculoesquelético en la EP de inicio tardío.
Recomendaciones para el manejo y el tratamiento musculoesquelético de la enfermedad de Pompe de inicio tardío
| Examen y evaluaciones físicas |
| Examen por un cardiológico y neumonólogo para determinar si el paciente puede iniciar un plan de rehabilitación física |
| Incluir en la evaluación una densitometría ósea, independientemente de la edad y/o uso de silla de ruedas. Realizar consulta con especialista en osteoporosis si se detectan anomalías. Utilizar, de ser necesario, vitamina D, calcio y bifosfonatos |
| Evaluar la marcha y el riesgo de caídas y fracturas óseas e implementar asistencia para la marcha si existe riesgo aumentado por osteopenia/osteoporosis. Puede incluir bastón, andador, o silla de ruedas según el trayecto a cumplir |
| Terapia física y ocupacional |
| Las actividades físicas a desarrollar deberían incluir una o algunas de las siguientes: caminata o cinta caminadora, ciclismo, ejercicio en pileta o nado, ejercicio aeróbico submáximo o de fortalecimiento muscular |
| Los principios a respetar son los mismos que para otras enfermedades neuromusculares, como por ejemplo: |
| – Evitar la fatiga por exceso de trabajo, desuso, ejercicios extenuantes o que involucren contracciones excéntricas. El ejercicio aeróbico debe ser submáximo |
| – Los ejercicios deben comenzar lentamente, permitiendo períodos de descanso, seguidos por un aumento gradual en la intensidad de leve a moderada |
| – Instruir al paciente para reconocer los síntomas de fatiga muscular e interrumpir el ejercicio antes de que estos se presenten, así como a vigilar la frecuencia cardíaca y la respiración en relación al ejercicio |
| – Realizar ejercicios posturales preventivos y enseñar las mejores posturas en la vida diaria con un fin ergonómico. Comenzar un plan temprano de elongaciones para minimizar el desarrollo de contracturas musculares y deformidades, y evitar posiciones inapropiadas. Incluir, de ser necesario, la utilización de férulas o valvas |
| – Evaluar la necesidad de intervenciones quirúrgicas en conjunto con neumonólogos y neuroortopedistas. Para la escoliosis, podría considerarse tratamiento quirúrgico si el ángulo de Cobb se encuentra entre 30° y 40°, dependiendo de la edad y condición respiratoria del paciente |
La debilidad de los músculos paravertebrales y del tronco son factores predisponentes al desarrollo de las deformaciones de la columna en la EP. La escoliosis es frecuente en los pacientes de inicio tardío y puede encontrársela hasta en un tercio de los casos. Es más común entre quienes comienzan con los síntomas en la niñez que en la vida adulta y en los pacientes en silla de ruedas42.
No existen trabajos o estudios respecto del tratamiento o manejo específico de la escoliosis en la EP, por lo que se siguen las pautas recomendadas en otras enfermedades neuromusculares.
El tratamiento quirúrgico suele ser el único verdaderamente útil para mejorar y prevenir la progresión de la deformidad espinal. La mayor experiencia en el manejo de la escoliosis en las ENM se tiene en distrofia muscular de Duchenne. En general, la cirugía se lleva adelante cuando el ángulo de Cobb se sitúa alrededor de 30 o 40°124-128.
Osteopenia en la enfermedad de PompeComo en otras ENM, la osteopenia es frecuente en la EP, tanto en niños como en adultos129. La condición ósea debe ser vigilada en estos pacientes a través de estudios de densitometría ósea, dual-energy X-ray absorptiometry10.
Aspectos gastrointestinales y nutricionalesTodos los pacientes con EP requieren una evaluación nutricional y gastroenterológica integrales desde el momento de realizado el diagnóstico. Deben investigarse los hábitos alimentarios, así como la calidad y la cantidad de alimentos ingeridos, la tolerancia y dificultades en la masticación y la deglución50.
Las formas infantiles presentan con frecuencia dificultad en la succión, sialorrea, macroglosia, debilidad lingual e hipotonía velopalatina y facial. Secundariamente, pueden aparecer trastornos en la masticación y la deglución10. Estas complicaciones pueden ocasionar déficits nutricionales, por lo que el crecimiento y el desarrollo de estos pacientes deben ser controlados periódicamente130. Las alteraciones citadas coadyuvan en la aparición de complicaciones secundarias, como microaspiraciones, neumopatías, infecciones, constipación pertinaz, déficits absortivos, osteopenia y osteoporosis, entre otros cuadros.
La debilidad de los músculos bulbares es un hecho frecuente en pacientes adultos con EP131. La disfunción digestiva también ha sido reportada, así como su mejoría con la TRE132. Las anomalías en la deglución en pacientes con EP pueden ser diagnosticadas mediante una evaluación videofluoroscópica. Este estudio identifica las deficiencias en las distintas fases de la deglución, como la fase oral y faríngea, en la que se puede apreciar residuos en la valécula o seno piriforme después de la deglución10.
La debilidad en los músculos de la lengua puede ser evaluada clínicamente43 y por imágenes106 en pacientes adultos con EP.
En ocasiones, puede ser necesario proteger al paciente del riesgo de aspiración por los trastornos deglutorios o la existencia de reflujo nasogástrico mediante el uso de sonda nasogástrica, asegurando así el aporte nutritivo.
Manejo nutricionalEs aconsejable que el plan nutricional sea manejado por un especialista que profundice en los hábitos alimentarios y nutricionales, además de dar indicaciones sobre la manipulación de alimentos y pautas de alimentación apropiadas. El programa nutricional se basa en la disminución de los hidratos de carbono y el aumento de aminoácidos como fuente energética10,111,133,134. También debe contemplar recomendaciones en la preparación y la elección de los alimentos, y en la frecuencia y el tamaño de las porciones.
Terapia de reemplazo enzimáticoHasta el año 2000, no hubo disponible ningún tratamiento específico para la EP, por lo que solo se aplicaban medidas de sostén. Algunas de las terapéuticas utilizadas, como la adrenalina y el glucagón, intentaban mejorar la degradación citosólica de glucógeno, pero no fueron exitosas135. Otras terapias mostraron solo efectos limitados en algunos individuos, como la alanina, las dietas ricas en proteínas, los aminoácidos de cadena ramificada y el betaagonista albuterol136-140. Existen, por último, terapias como el trasplante de médula ósea y de corazón, sobre las cuales se tiene una experiencia muy limitada10,141.
En la actualidad, la TRE es el único tratamiento específico para la EP. La TRE abastece de forma exógena la enzima faltante a través de infusiones por vía intravenosa repetidas. La AGA humana recombinante (AGAhr) (Myozyme®; Genzyme Corporation) ha demostrado ser eficaz en el tratamiento de los pacientes con EP de inicio precoz y tardío10,21,22,142-145. La dosis recomendada es de 20mg/kg de peso corporal administrada una vez cada 2 semanas por vía endovenosa. Se probaron dosis mayores, de 40mg/kg de peso cada 2 semanas, pero no demostraron un beneficio claro por sobre la dosis habitual, y parecen asociarse con mayor frecuencia a eventos adversos22,143.
Varios ensayos en pacientes con EP de inicio infantil mostraron que la TRE prolonga la sobrevida en forma significativa, disminuye la cardiomegalia y mejora la función de los músculos cardíaco y esquelético, y el desarrollo motor21,145-152. En la gran mayoría de los casos, la respuesta sobre el músculo cardíaco fue muy buena, de forma independiente de cuál era la fase de la enfermedad al inicio de la TRE. Los pacientes tratados adquirieron en ocasiones habilidades motoras y funcionales, como la deambulación independiente, que son muy inusuales en pacientes no tratados143. La utilización sostenida en el tiempo de la TRE ha llevado a la aparición de un nuevo fenotipo de pacientes con EP infantil y sobrevida prolongada, con un patrón específico de compromiso153.
En la EP de inicio tardío, el objetivo principal de la TRE es la prevención de la pérdida adicional de la función muscular22. Si bien la experiencia en adultos es más limitada, los datos indican que la AGAhr tiene un efecto positivo sobre la historia natural de la EP y sobre los procesos que llevan a la insuficiencia respiratoria y el deterioro de la deambulación22,154. En el conjunto de los distintos estudios realizados hasta el año 2012, al menos 2 tercios de los pacientes se estabilizaron o mejoraron sus niveles de CK y la función respiratoria y/o muscular luego de la TRE90. El beneficio en la deambulación y la estabilización en la función ventilatoria parecen prolongarse al menos hasta la semana 104 de infusión continuada88. Un estudio reciente de una población de 283 pacientes con EP ha demostrado el impacto positivo de la TRE en la sobrevida de estos pacientes155. Los resultados de algunos estudios indican un mayor beneficio cuando la TRE se instaura en fase precoz y en pacientes cuya situación clínica basal está mejor conservada151,156, pero también se ha observado mejoría o estabilización de la enfermedad en pacientes con estadios avanzados o muy discapacitados por la enfermedad157.
El tratamiento con TRE para la EP de inicio tardío fue aprobado en el año 2006 por la Agencia Europea de Medicamentos y en el 2010 por la FDA.
Recomendaciones para el inicio del tratamiento de la enfermedad de Pompe de inicio tardíoTeniendo en cuenta el análisis de distintos reportes de la literatura y la evidencia disponible sobre la indicación de TRE en pacientes con EP de inicio tardío23, se pueden distinguir los siguientes grupos (véanse también las tablas 4 y 5):
- 1.
Individuos asintomáticos sin signos objetivos, cuyo diagnóstico fue alcanzado por estudios de laboratorio (por tener familiares afectados o mediante métodos de cribado): no hay evidencia bibliográfica que demuestre que el tratamiento con TRE de este grupo retrase la aparición de la enfermedad, por lo que no existe sustento para recomendar el inicio del mismo. Los sujetos en esta condición deben ser examinados cada 6 meses con evaluación de la fuerza muscular (en lo posible cuantitativa) y función respiratoria. La TRE debería iniciarse ante la aparición de síntomas asociados a debilidad muscular o reducción de la CVF que pueda ser atribuida a la enfermedad, es decir, solo cuando se encuentren los primeros signos objetivos de EP. Es importante tener en cuenta que si se detecta la presencia de EP en un determinado sujeto, se recomienda encarar la investigación diagnóstica en los hermanos158.
- 2.
Pacientes asintomáticos, con signos objetivos: este grupo incluye a pacientes con debilidad muscular o reducción de la CVF que puedan ser atribuidas a la enfermedad, y deberían iniciar tratamiento con AGAhr.
- 3.
Pacientes sintomáticos: la TRE con AGAhr se recomienda para los pacientes sintomáticos que tengan debilidad muscular demostrable en el examen físico o reducción de los parámetros pulmonares en las pruebas de función respiratoria. En caso contrario, puede considerarse la TRE si el paciente tiene dificultades en la realización de las actividades de la vida diaria que puedan ser atribuidas a la enfermedad.
Recomendaciones para el inicio del tratamiento con terapia de reemplazo enzimático de la enfermedad de Pompe basadas en el estadio y la severidad
| Condición | Recomendación |
| Forma infantil de la EP | Debe iniciarse el tratamiento tan rápido como sea posible una vez realizado el diagnóstico |
| Individuos asintomáticos sin signos objetivos (diagnóstico bioquímico/genético) | Deben ser examinados cada 6 meses con evaluación de la fuerza muscular (en lo posible cuantitativa) y función respiratoria. La TRE debería iniciarse ante la aparición de síntomas asociados a debilidad muscular o reducción de la CVF correlacionable con la enfermedad |
| Pacientes asintomáticos, con signos objetivos (EP de inicio tardío) | Debería iniciarse TRE si el paciente presenta debilidad muscular detectable o reducción de la CVF que puedan ser atribuidas a la enfermedad |
| Pacientes sintomáticos (EP de inicio tardío) | Debería iniciarse TRE si se encuentra debilidad muscular detectable o reducción de la CVF relacionadas con la enfermedad. En caso contrario, puede considerarse la TRE si el paciente tiene dificultades en la realización de las actividades de la vida diaria que puedan ser atribuidas a esa causa |
CVF: capacidad vital forzada; EP: enfermedad de Pompe; TRE: terapia de reemplazo enzimático.
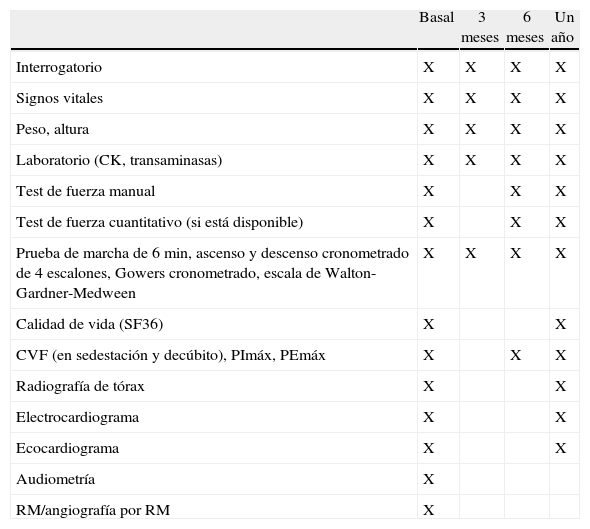
Protocolo de evaluaciones en el paciente con enfermedad de Pompe del adulto previa y durante la terapia de reemplazo enzimático
| Basal | 3 meses | 6 meses | Un año | |
| Interrogatorio | X | X | X | X |
| Signos vitales | X | X | X | X |
| Peso, altura | X | X | X | X |
| Laboratorio (CK, transaminasas) | X | X | X | X |
| Test de fuerza manual | X | X | X | |
| Test de fuerza cuantitativo (si está disponible) | X | X | X | |
| Prueba de marcha de 6 min, ascenso y descenso cronometrado de 4 escalones, Gowers cronometrado, escala de Walton-Gardner-Medween | X | X | X | X |
| Calidad de vida (SF36) | X | X | ||
| CVF (en sedestación y decúbito), PImáx, PEmáx | X | X | X | |
| Radiografía de tórax | X | X | ||
| Electrocardiograma | X | X | ||
| Ecocardiograma | X | X | ||
| Audiometría | X | |||
| RM/angiografía por RM | X |
CVF: capacidad vital forzada; CK: creatincinasa; PEmáx: presión espiratoria máxima; PImáx: presión inspiratoria máxima; RM: resonancia magnética.
Una vez que se inició la TRE, este tratamiento es a largo plazo, tal vez durante toda la vida. Vale la pena destacar que la respuesta individual a la TRE puede variar debido a distintos factores, como el desarrollo de anticuerpos específicos contra la AGAhr, la edad de presentación, la tasa de progresión de la enfermedad, el tipo de fibra muscular, la presencia de defectos en la autofagia y el genotipo subyacente8,10. La discontinuación de la TRE debe considerarse si existen reacciones adversas serias que pongan en peligro la vida del paciente o complicaciones graves derivadas de la instauración de la misma.
Desarrollo de anticuerpos específicosEl desarrollo de anticuerpos anti-AGAhr es más frecuente en pacientes con ausencia de AGA endógena, que carecen de material inmunológicamente reactivo (CRIM-negativos, por su sigla en inglés, cross-reactive immunologic material). Esto tendría un impacto negativo en el pronóstico de los pacientes con EP infantil8.
Con el objetivo de disminuir el efecto deletéreo de la formación de anticuerpos contra AGAhr, se ha intentado mejorar la tolerancia inmunológica, para lo cual existen ensayos en curso con modelos de ratones knockout para AGA159,160. Se han obtenido logros en niños CRIM negativos utilizando un régimen inductor de tolerancia que incluye el uso de anticuerpos monoclonales anti-CD20 (rituximab), metotrexato y gammaglobulina por vía intravenosa161,162. Como, por lo general, la TRE por sí sola tiene pobres resultados en los pacientes CRIM negativos, es importante discutir este eventual panorama con los padres de los niños afectados.
Efecto de la terapia de reemplazo enzimático en el músculo esqueléticoLa mejor respuesta de la TRE sobre el músculo esquelético se ha observado en pacientes tratados de forma temprana, antes de un daño muscular irreversible. No obstante, esta respuesta ha sido más errática que la del músculo cardíaco. Esto podría deberse a que si bien la captación enzimática en ambos tipos musculares está mediada por receptores de manosa-6-fosfato, el músculo esquelético tendría muchos menos receptores que el corazón163,164, lo que podría redundar en una menor captación de la enzima. Existen otras teorías que tienden a explicar la menor efectividad de la TRE sobre el músculo esquelético163,164. A esto se debe añadir que dicho músculo constituye aproximadamente el 40% de la masa corporal total, por lo que el volumen a tratar es mucho mayor que el del músculo cardíaco.
Reacciones adversas a la terapia de reemplazo enzimáticoLa TRE es, en general, bien tolerada y la mayoría de los eventos adversos son leves o moderados90. Sin embargo, no está exenta de complicaciones potenciales. Los pacientes CRIM negativos, o aquellos que presentan una enzima nativa con diferencias significativas con la enzima recombinante administrada, pueden producir anticuerpos contra la enzima exógena, lo que reduce su eficacia y puede ocasionar reacciones adversas ante la infusión. Muchos pacientes también desarrollan tolerancia con el tiempo.
A pesar de que la mayoría de las reacciones a la infusión son, en general, fáciles de tratar, en algunos casos puntuales se han observado reacciones anafilácticas peligrosas, alergia, fiebre y respuestas autoinmunes22,154. Estas complicaciones pueden aparecer durante el tratamiento o en horas posteriores al mismo21,143.
Las reacciones anafilácticas son una complicación potencialmente grave del tratamiento con cualquier proteína humana recombinante, y se observan en alrededor de un 5% de los pacientes con EP de aparición tardía. Los pacientes tratados con AGAhr que desarrollen títulos altos de anticuerpos deben ser monitoreados en forma estrecha53, con un equipo médico siempre disponible durante la infusión. El tratamiento de pacientes con EP infantil debe hacerse en centros especializados con adecuada experiencia.
Si bien la EP no es una nueva entidad, la ciencia está hoy comenzando a conocer mejor algunos de sus mecanismos y cascadas fisiopatológicas, por lo que lo que actualmente sabemos de ella puede modificarse con el tiempo. Nuevas terapias están en desarrollo y las actuales indicaciones deberán ajustarse según la evolución de esos conocimientos.
Conflicto de interesesLos Dres. Dubrovsky, Ferradás, Fulgenzi, Lubienicki, Mesa, Reisin, Rugiero y Zgaga declaran haber recibido honorarios por conferencias y/o clases por parte de laboratorios Genzyme. Los Dres. Fulgenzi, Mazia, Mesa, Rugiero y Zgaga declaran haber recibido viáticos para asistencia a Congresos y/o eventos científicos de parte de laboratorios Genzyme. El Dr. Reisin declara haber recibido becas para viajes de estudios de parte de laboratorios Genzyme. Los Dres. Dubrovsky y Rugiero declaran haber recibido subsidios de investigación de parte de laboratorios Genzyme. El Dr. Dubrovsky es miembro del Global Advisory Board for Pompe Disease del laboratorio Genzyme.
Los Dres. de Vito y Rugiero declaran haber recibido honorarios por conferencias por parte de laboratorios Sanofi. Los Dres. Fulgenzi, Mazia y Rugiero declaran haber recibido viáticos para asistencia a Congresos y/o eventos científicos de parte de laboratorios Sanofi. El Dr. Rugiero declara haber recibido subsidios de investigación por parte de laboratorios Sanofi.
Las Dras. Lubienicki y Taratuto declaran haber recibido honorarios por conferencias de parte de laboratorios Genzyme y/o Sanofi. El Dr. Guelbert declara haber recibido honorarios por conferencias en Jornadas y Congresos relacionados con enfermedades congénitas del metabolismo durante el año 2013. La Dra. Lubienicki declara haber recibido subsidios para investigación de parte de laboratorios Genzyme y/o Sanofi. El licenciado Corderi declara haber recibido viáticos o subsidios de investigación u honorarios por conferencias de parte de laboratorios Genzyme y/o Sanofi.
Los Dres. Monjes y Pesquero declaran no presentar potencial conflicto de interés.
Los Dres. Amartino, Carlés, Fainboim, Schenone y Szlago no han declarado la existencia de potencial conflicto de interés hasta la fecha.
