




La unión neuromuscular es el blanco de diferentes trastornos autoinmunes, dirigidos a distintos canales indispensables para la transmisión de señales neuromusculares, como los canales de calcio y potasio activados por voltaje en la membrana presináptica, y los receptores de acetilcolina en la membrana postsináptica. Los anticuerpos contra estos canales provocan un grupo heterogéneo de enfermedades, como el síndrome de Lambert-Eaton, el síndrome de Isaacs y la Miastenia Gravis. Aunque estas canalopatías comparten algunas características comunes, difieren en las características clínicas, el perfil de anticuerpos, las características neurofisiológicas y los tratamientos. Todos estos trastornos pueden tener tumores asociados.
Objetivo principalConocer la utilidad y las limitaciones de la determinación de los anticuerpos relacionados con estas entidades.
MétodosRevisión bibliográfica en las bases de datos PubMed-NCBI, SciELO y LILACS. Se describen antígeno, anticuerpo, mecanismo patogénico, frecuencia de su hallazgo, métodos de determinación con su sensibilidad y especificidad, valores de referencia y utilidad en la práctica, clínica y la electrofisiología.
ConclusionesLos anticuerpos dirigidos a antígenos pre y postsinápticos causan enfermedades cuya manifestación común es la fluctuación de la fuerza muscular. La positividad de los anticuerpos tiene una certeza diagnóstica muy variable, dependiente de diversos factores. Los anticuerpos contra proteínas intracelulares no tienen un rol patogénico dilucidado y algunos son utilizados como marcadores de severidad y pronóstico. Las características clínicas y electrofisiológicas contribuyen a definir fehacientemente el diagnóstico. La autoinmunidad de origen paraneoplásico siempre debe ser descartada. La factibilidad de tratamiento de estos trastornos exige precocidad en el diagnóstico.
Some neuromuscular junction proteins, such as voltage-gated calcium and potassium channels on the presynaptic membrane and acetylcholine receptors on the postsynaptic membrane, are the antigenic target of different autoimmune disorders. Antibodies against these channels cause diseases, among which Lambert-Eaton myasthenic syndrome, Isaacs syndrome (pre-synaptic) and myasthenia gravis (post-synaptic) are the most common.
MainTo describe the usefulness and limitations of determining neuromuscular junction antibodies.
MethodsLiterature review in PubMed-NCBI, SciELO and LILACS, outlining antibodies and their target antigens, frequency of their finding, mechanism of action, clinical and electrophysiological characteristics, methods of determination with sensitivity and specificity, reference values and practical utility.
ConclusionsAt the neuromuscular junction, signal transmission depends on pre- and postsynaptic channels. Antibodies directed Antibodies directed to pre- and postsynaptic antigens cause diseasewhose common manifestation is fluctuation of muscle strength. The positivity of antibodies have a highly variable diagnostic certainty, dependent on diverse factors.Antibodies against intracellular proteins do not have an elucidated pathogenic role and some are used as markers of severity and prognosis. The characteristics clinical and electrophysiological studies help to reliably define the diagnosis. The autoimmunity of paraneoplastic origin must always be ruled out. The feasibility of treatment of these disorders requires early diagnosis.
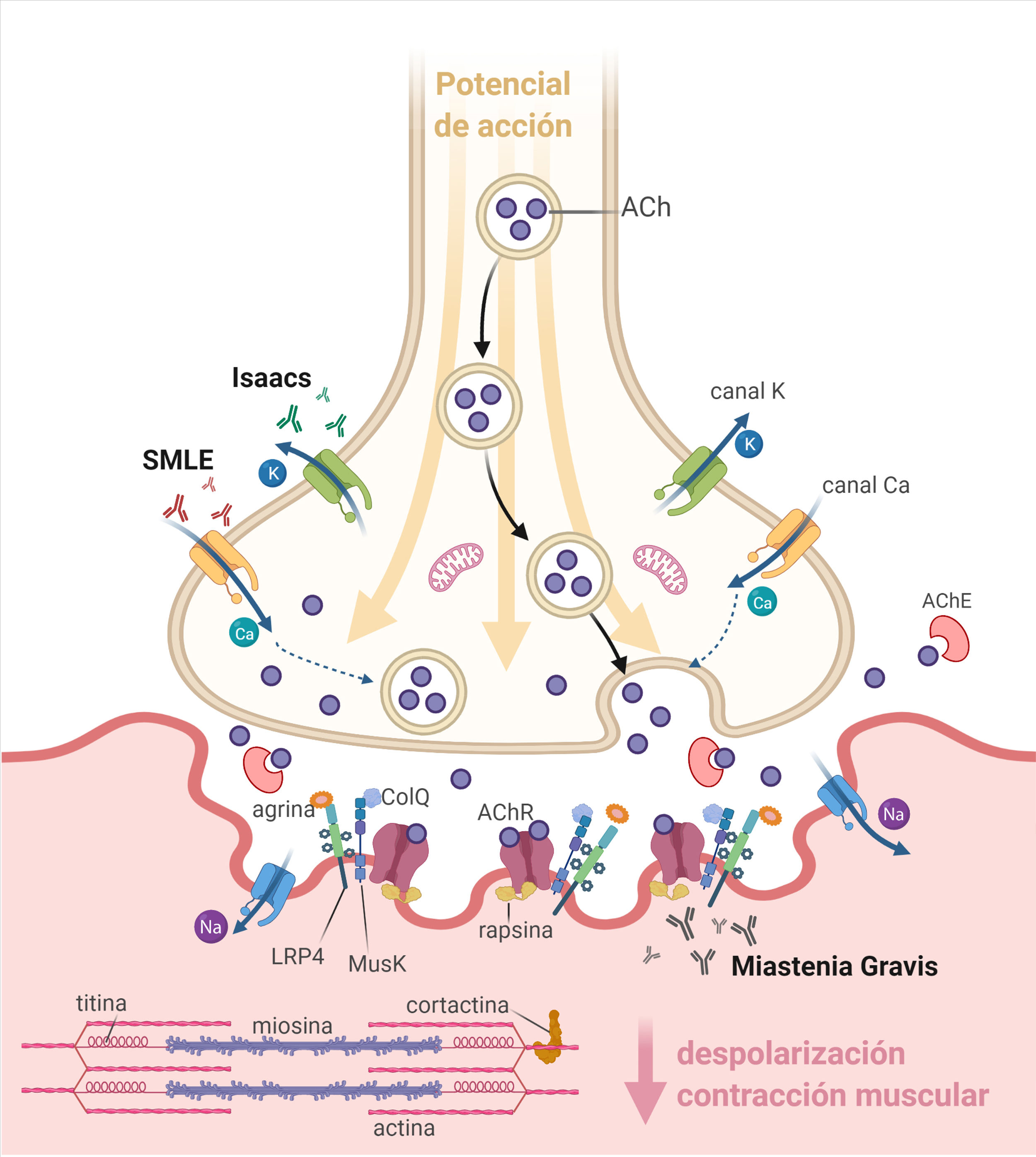
La unión neuromuscular (UNM) está constituida por el terminal axonal (presináptico), la hendidura sináptica y la fibra muscular (postsináptica) (fig. 1). En la membrana presináptica se encuentran los canales de calcio dependientes de voltaje (VGCC) y los canales de potasio dependientes de voltaje (VGKC). En los pliegues primarios de la membrana postsináptica se encuentran los receptores nicotínicos de acetilcolina (AChR), la proteína tirosina cinasa específica de músculo (MuSK), la proteína 4 asociada con el receptor de lipoproteínas de baja densidad (LRP4) y otras proteínas, mientras que en los pliegues secundarios existe una alta densidad de canales de sodio dependientes de voltaje (VGSC)1,2.

La liberación espontánea de acetilcolina (ACh) desde el terminal axonal genera en el sarcolema un potencial de placa (EPP) en miniatura. Tras la estimulación del nervio motor se produce la apertura de los VGCC y la liberación de una cantidad suficiente de ACh como para que se genere un EPP cuya amplitud sobrepasa el umbral de apertura de los VGSC y origina un potencial de acción muscular que se propaga e induce la contracción de la fibra muscular1,2.
Las enfermedades autoinmunes de la UNM se caracterizan por la presencia de autoanticuerpos dirigidos a moléculas de la membrana pre o postsináptica que, por diversos mecanismos, impiden que la amplitud del EPP llegue al umbral de disparo del potencial de acción muscular, lo cual se expresa clínicamente como debilidad fluctuante y fatigabilidad1,2.
ObjetivosObjetivo principalEl propósito de este trabajo de revisión es conocer la utilidad y las limitaciones de la determinación de anticuerpos en las enfermedades más frecuentes de la neurotransmisión.
Objetivos secundarios- -
Describir los anticuerpos y sus blancos antigénicos localizados en la membrana pre y postsináptica e intracelulares.
- -
Conocer el mecanismo patogénico de dichos anticuerpos.
- -
Revisar la frecuencia, los valores de referencia, la sensibilidad y la especificidad de los métodos de determinación de cada anticuerpo.
- -
Mencionar las manifestaciones clínicas, paraneoplásicas y electrofisiológicas que contribuyen al diagnóstico en cada entidad.
Se consultaron las bases de datos PubMed-NCBI, SciELO y LILACS utilizando las palabras clave: unión neuromuscular, anticuerpos, miastenia gravis, Lambert-Eaton, síndrome de Isaacs, hiperexcitabilidad del nervio periférico. Con base en la literatura revisada se realizó una descripción de las características clínicas y electrofisiológicas de cada entidad, así como de cada antígeno, anticuerpo, mecanismo patogénico, frecuencia de su hallazgo, métodos de determinación con su sensibilidad y especificidad, valores de referencia y utilidad en la práctica. En relación con la utilidad de medir los anticuerpos, también se expresa la experiencia de los autores.
ResultadosEnfermedades con anticuerpos dirigidos a estructuras presinápticas (tabla 1)Síndrome miasténico de Lambert-EatonEl síndrome miasténico de Lambert-Eaton (SMLE)3 es el resultado de un ataque autoinmune contra los VGCC tipo P/Q de la membrana presináptica, involucrados en la liberación de ACh4–7. Fueron Fukunaga et al. los primeros en plantearlo en 1983. Subsecuentes estudios por Engel, Vincent y Newsom-Davis demostraron la patogenia autoinmune subyacente8; otros investigadores confirmaron una reducción de la amplitud del EPP en miniatura, EPP y de la liberación de ACh en el SMLE9.
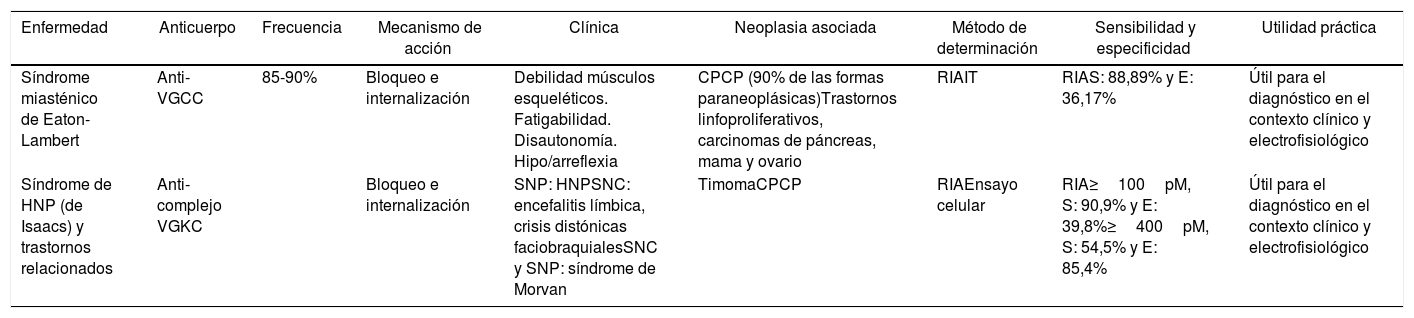
Enfermedades presinápticas
| Enfermedad | Anticuerpo | Frecuencia | Mecanismo de acción | Clínica | Neoplasia asociada | Método de determinación | Sensibilidad y especificidad | Utilidad práctica |
|---|---|---|---|---|---|---|---|---|
| Síndrome miasténico de Eaton-Lambert | Anti-VGCC | 85-90% | Bloqueo e internalización | Debilidad músculos esqueléticos. Fatigabilidad. Disautonomía. Hipo/arreflexia | CPCP (90% de las formas paraneoplásicas)Trastornos linfoproliferativos, carcinomas de páncreas, mama y ovario | RIAIT | RIAS: 88,89% y E: 36,17% | Útil para el diagnóstico en el contexto clínico y electrofisiológico |
| Síndrome de HNP (de Isaacs) y trastornos relacionados | Anti-complejo VGKC | Bloqueo e internalización | SNP: HNPSNC: encefalitis límbica, crisis distónicas faciobraquialesSNC y SNP: síndrome de Morvan | TimomaCPCP | RIAEnsayo celular | RIA≥100pM, S: 90,9% y E: 39,8%≥400pM, S: 54,5% y E: 85,4% | Útil para el diagnóstico en el contexto clínico y electrofisiológico |
CPCP: carcinoma de pulmón de células pequeñas; E: especificidad; HNP: hiperexcitabilidad del nervio periférico; IT: inmunotransferencia; RIA: radioinmunoanálisis; S: sensibilidad; SNC: sistema nervioso central; SNP: sistema nervioso periférico; VGCC: canal de calcio dependiente de voltaje; VGKC: canal de potasio dependiente de voltaje.
El SMLE es paraneoplásico en el 60% de los casos, principalmente asociado a carcinoma de pulmón de células pequeñas (CPCP). Una relación con HLA B8DR3 evidencia una predisposición genética a la autoinmunidad5,10.
En la forma paraneoplásica la edad media de inicio es de 60 años con predominio masculino, mientras que en el SMLE primario hay 2 edades pico: a los 35 y a los 60 años. La distribución por edad y sexo en el SMLE primario es similar a la registrada para miastenia gravis (MG)11. Inicia insidiosamente, la debilidad muscular es fluctuante y con una distribución diferente a la de la MG, dado que el tronco y los miembros inferiores están más involucrados. La afectación ocular y el compromiso de los músculos respiratorios son inusuales. Frecuentemente hay compromiso parasimpático, con xerostomía, estreñimiento, impotencia sexual e hipotensión ortostática12,13. Los reflejos están deprimidos, fundamentalmente en los miembros inferiores; al igual que la fuerza, mejoran inmediatamente luego de una contracción voluntaria, esto se evidencia clínica y electrofisiológicamente y se conoce como «fenómeno de facilitación».
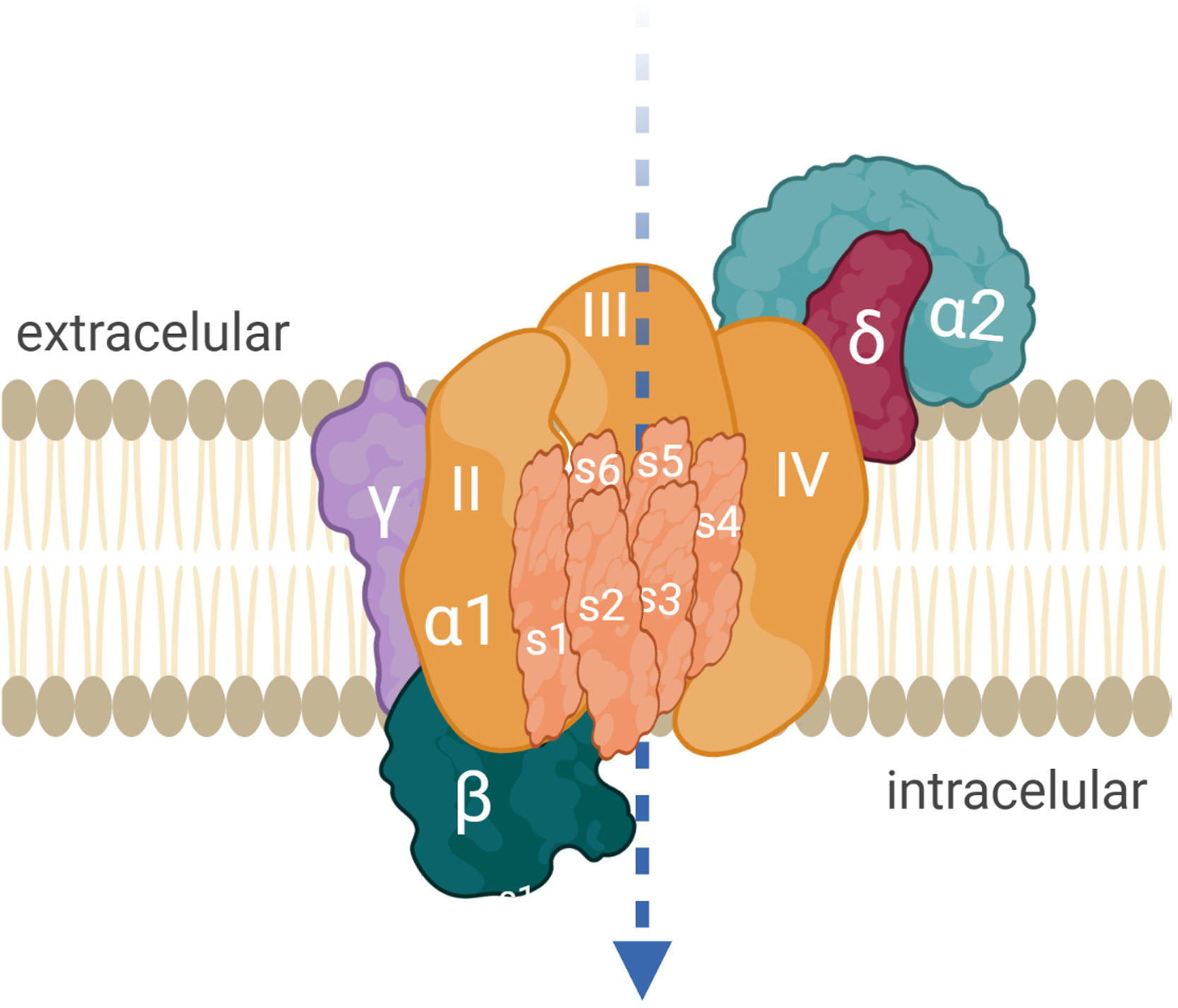
AntígenoEl VGCC permite el influjo celular de calcio cuando el potencial de acción nervioso despolariza el terminal axonal. El catión facilita la fusión de la vesícula que almacena ACh al neurolema presináptico y su liberación al espacio sináptico. El canal está constituido por proteínas oligoméricas con una subunidad principal α1 y varias subunidades reguladoras o auxiliares14. La subunidad α1 del canal P/Q tiene 4 dominios (i-iv) y cada uno tiene 6 segmentos transmembrana (S1-S6). S4 es sensor de voltaje, S5 y S6 son sensores de calcio (fig. 2). Hay 5 tipos de VGCC: L, P/Q, N, R, T. La presencia de VGCC P/Q en células de Purkinje explica la ataxia en pacientes con SMLE. Los de tipo N son relevantes en el sistema nervioso autónomo12.
Anticuerpo anticanal de calcio dependiente de voltaje
Los anti-VGCC están presentes en el SMLE primario y paraneoplásico y en la degeneración cerebelosa, fundamentalmente paraneoplásica5,12. Son IgG dirigidos contra la subunidad α1 del VGCC tipo P/Q14. El 30-40% también tiene anticuerpos contra los canales de tipo N y el 25% contra los de tipo L5. Los títulos no se correlacionan con la severidad de la enfermedad. Los VGCC de tipo P/Q se expresan en células de CPCP y esto origina una reacción cruzada con los VGCC presinápticos. El diagnóstico de SMLE suele preceder al diagnóstico de la neoplasia. Los anti-VGCC pueden detectarse en el 3-5% de los pacientes con CPCP sin síntomas neurológicos15.
Ocasionalmente pueden detectarse anticuerpos que se ligan a sinaptotagmina (involucrada en la fusión de vesículas)16 y a SOX-1, proteína relacionada con la génesis tumoral. Este último es positivo en el 67% de los pacientes con SMLE-CPCP17.
Mecanismo de acciónLos anti-VGCC bloquean el influjo de calcio al terminal presináptico, principalmente por la afinidad hacia los segmentos S5-S6 extracelulares de los dominios ii, iii y iv, de la subunidad α1 (fig. 2). Los anticuerpos son divalentes, lo cual implica que ambos brazos FAB de la IgG se unen al blanco antigénico y provocan la internalización de los VGCC18. No activan el complemento16. El resultado es la pérdida de canales de calcio funcionalmente disponibles, la reducción del influjo de calcio y finalmente de la liberación de cuantos de ACh. La traducción electrofisiológica es la reducción de la amplitud de los EPP en miniatura y de los EPP, y clínicamente, la debilidad muscular fluctuante con fenómeno de facilitación. La afección de estos receptores también ocurre en neuronas autonómicas.
Los anticuerpos contra sinaptotagmina interfieren en la fusión de las vesículas de ACh al neurolema, paso previo a su liberación16.
FrecuenciaLos anti-VGCC se detectan en el 85-90% de los pacientes con SMLE y hasta en el 100% de los pacientes con SMLE y CPCP19.
Métodos de determinaciónMediante radioinmunoanálisis (RIA) de VGCC combinado con ω-conotoxina radiomarcada con 125I. Se utilizan pruebas adicionales para aumentar la especificidad, como la inmunotransferencia20.
Especificidad y sensibilidadAnti-VGCC por RIA tiene una sensibilidad del 88,89% y una especificidad de 36,17%, esta última es directamente proporcional al título de anticuerpos detectado. Títulos≥1nmol/L presentan enfermedad neurológica autoinmune con mayor frecuencia que los pacientes con valores intermedios (0,10-0,99nmol/L) o bajos (0,03-0,10nmol/L)20.
Utilidad en la prácticaDebido a la baja especificidad del RIA, la presencia de anti-VGCC debe interpretarse con cautela, fundamentalmente con títulos bajos e intermedios, jerarquizando la clínica y la electrofisiología.
Se debe considerar que el diagnóstico de SMLE puede preceder a la detección de una neoplasia, fundamentalmente CPCP.
No hay relación entre la positividad de VGCC o SOX-1 y la sobrevida20.
Los anticuerpos dirigidos contra el AChR (AChRA) son encontrados en un ∼5-10%20, por lo tanto, el diagnóstico diferencial con MG puede resultar complejo si no contamos con la electrofisiología.
Síndrome de Isaacs (hiperexcitabilidad de nervio periférico) y otros trastornos relacionados con los canales de potasio dependientes de voltajeLos anticuerpos dirigidos contra el complejo VGKC se asocian con 4 entidades: encefalitis límbica (EL), crisis distónicas faciobraquiales (DFB), síndrome de hiperexcitabilidad de nervio periférico (HNP), neuromiotonía o síndrome de Isaacs y síndrome de Morvan (SM). Este último se caracteriza por encefalitis, con amnesia, insomnio, disautonomía e HNP21. Se ha publicado asociación con neoplasia hasta en un 30% de los casos22, siendo con un timoma la más frecuente.
La HNP se asoció por primera vez con anticuerpos contra el VGKC en 199523. En 2001, los anti-VGKC fueron hallados en el SM24 y en la EL25, y en el 2008 en las crisis DFB26. En 2010 se describió que estos anticuerpos se ligan también a otras proteínas que conforman un complejo con el canal de potasio propiamente dicho (Kv). Desde entonces se reconoce a todo el complejo VGKC como potencial blanco antigénico22.
ClínicaEn esta revisión nos enfocaremos en las manifestaciones neuromusculares.
El síndrome de HNP inicia entre los 15-60 años, la mayoría antes de los 40. Consiste en contracción muscular involuntaria, ondulante y persistente en reposo, dificultad en la relajación (pseudomiotonía), calambres, fasciculaciones, mioquimias que continúan durante el sueño y debilidad muscular. Puede observarse hipertrofia muscular, dolor e hiperestesia. La disfunción autonómica es frecuente, con hiperhidrosis, rubor, vasodilatación, piloerección y dolor abdominal21,27. El electromiograma característico registra descargas agrupadas en dupletes, tripletes o multipletes, con frecuencia de descarga alta, fibrilaciones y fasciculaciones27,28. Otras enfermedades autoinmunes, como la MG, y neoplasias como el timoma y el CPCP pueden estar presentes.
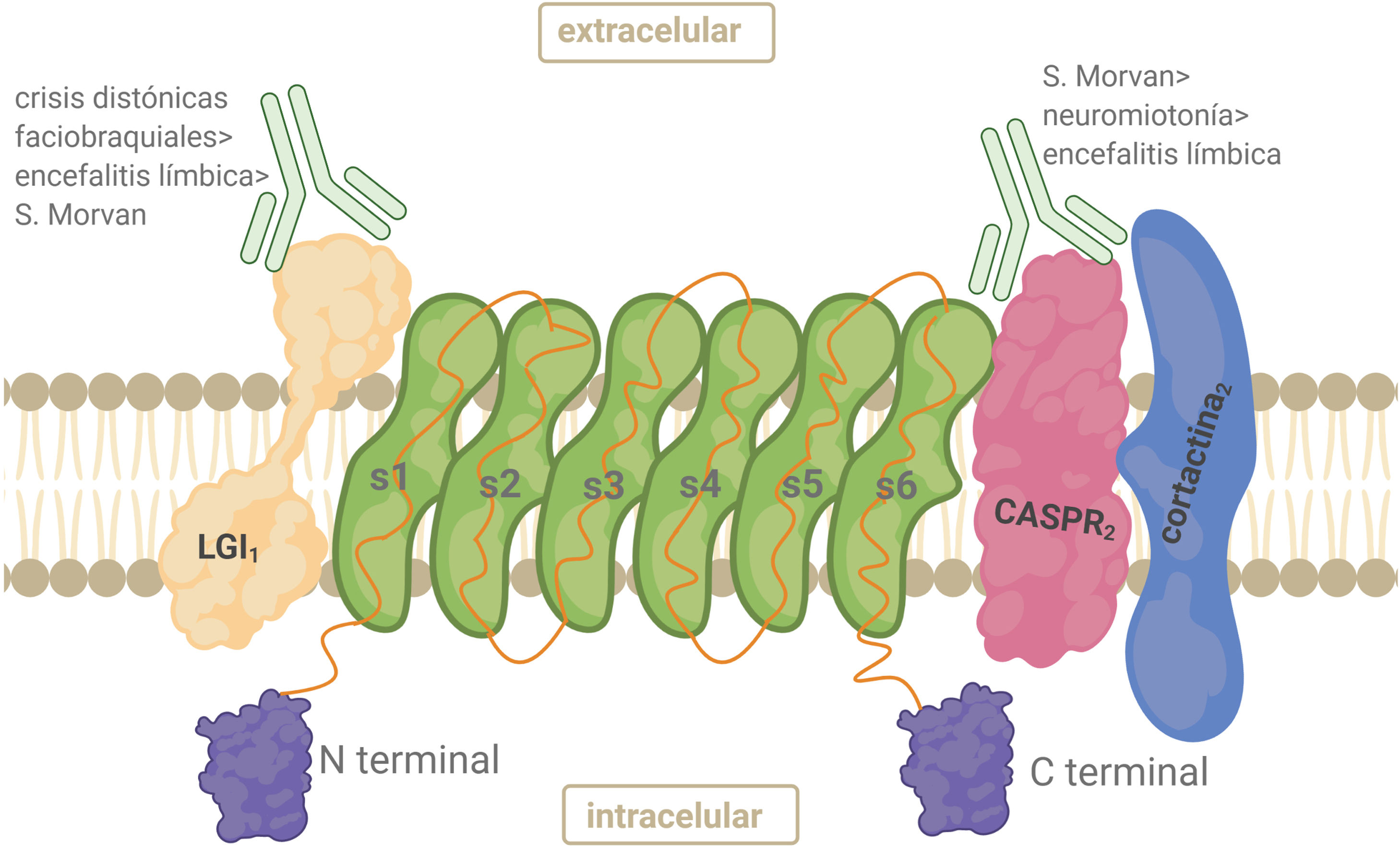
AntígenoEl complejo VGKC está constituido por el Kv, la proteína 1 inactivada de glioma rica en leucina (LGI1), la proteína 2 asociada a contactina (CASPR2) y contactina-2. LGI1 se expresa principalmente en el SNC. LGI1 forma complejos con Kv1 en las regiones yuxtaparanodal, en el segmento inicial del axón y en las terminales sinápticas. Es una glucoproteína secretada que se une a la superficie celular. La parte N-terminal contiene 3 repeticiones ricas en leucina que funcionan como dominio de unión a proteínas22 (fig. 3). CASPR2 es un miembro de la superfamilia de neurexinas (proteínas transmembrana) implicadas en las interacciones célula-célula dentro del sistema nervioso. CASPR2 sirve como andamio para Kv1.1/ Kv1.2 en la región yuxtaparanodal tanto en el SNP como en el SNC22 (fig. 3).

Los canales iónicos se clasifican en 12 grupos (Kv1-12), se activan por la despolarización y el eflujo de potasio repolariza la membrana26.
Anticuerpos anticomplejo canal de potasio dependiente de voltajeLos pacientes con HNP y SM tienen predominantemente anticuerpos contra CASPR2, mientras que los anti-LGI1 predominan en DFB y EL22,26,27. Los anticuerpos dirigidos a las subunidades kv1 y a contactina-2 son infrecuentes. Algunos anticuerpos podrían unirse a antígenos del complejo VGKC aún desconocidos, dando un resultado positivo para anti-VGKC pero negativo para anticuerpos LGI1 y CASPR229.
Anti-LGI1 son de subtipo IgG1 y principalmente IgG4. Los anticuerpos anti-CASPR2 son predominantemente del subtipo IgG1.
Mecanismo patogénicoEl mecanismo de estos anticuerpos es parcialmente conocido. Las IgG de los pacientes con HNP se ligan a las proteínas del complejo VGKC, y reducen el número de VGKC sin activación del complemento15. Bloquean el canal iónico de manera similar a las aminopiridinas23. Los anticuerpos contra LGI1 y CASPR2 disminuyen la expresión de VGKC30. La atenuación del flujo de potasio a través de la membrana axonal reduce crónicamente el potencial de membrana de reposo, afectando su repolarización con la consecuente hiperexcitabilidad.
FrecuenciaLos anticuerpos anti-CASPR2 se detectan en el 70% de los pacientes con SM, coexistiendo con anti-LGI1 y en el 30% de los pacientes con HNP, siendo en este último los anticuerpos predominantes. Un 40% de los casos de HNP no tiene blanco antigénico definido31–33. Anti-LGI1 está presente en un 95% de los casos de crisis DFB y en un 80% de los casos de EL. Son infrecuentes en HNP32. En LCR no suelen detectarse anticuerpos en HNP, mientras que sí en EL y SM.
Métodos de determinaciónSe emplea RIA con 125I-DTX, un ligando específico de ciertos subtipos Kv133. Los sueros positivos se analizan para LGI1, contactina-2, CASPR2 y Kv1 usando ensayo con células transfectadas y fluorescencia. Las determinaciones de LCR de rutina son normales o muestran hiperproteinorraquia leve. Su estudio puede ayudar a excluir otros trastornos25.
Valores de referenciaLos resultados se calculan como picomolar de 125I-DTX precipitado (valores normales<100pM). Títulos entre 100 y 400pM se encuentran en el 35% de las personas mayores, en el 40% de los pacientes con HNP y en algunos casos de SM. Títulos>400pM son consistentemente registrados cuando hay compromiso del SNC, como en la EL25.
Sensibilidad y especificidadLa sensibilidad y especificidad de los anti-VGKC con un título≥100pM es de 90,9 y 39,8% para el diagnóstico de HNP, respectivamente, mientras que con un título≥400pM la sensibilidad es del 54,5% y la especificidad aumenta al 85,4%34. Los títulos bajos (100-400pM) se pueden encontrar en pacientes con neoplasias y enfermedades neurodegenerativas. Títulos≥400pM se asocian con EL25.
Utilidad en la prácticaLa determinación de estos anticuerpos tiene relevancia en un contexto clínico y electrofisiológico. La especificidad del diagnóstico tiene relación con el título sérico hallado. Además, se puede determinar específicamente anti-LGI1 cuando la clínica es compatible con EL y DFB, y anti-CASPR2 cuando el paciente presenta HNP y timoma.
La positividad de anti-VGKC obliga a descartar neoplasias, fundamentalmente de timo.
Enfermedades con anticuerpos dirigidos a estructuras postsinápticas (tabla 2): Miastenia GravisLa posibilidad de que la MG fuera autoinmune fue propuesta por Nastuk et al.35. Simpson36 planteó la hipótesis de un anticuerpo contra un receptor de la UNM. Patrick y Lindstrom demostraron que la inmunización contra los AChR inducía una forma de «MG autoinmune experimental»37. El AChRA fue detectado por inmunoprecipitación con 125I-α-bungarotoxina38. Los cambios fisiopatológicos de la UNM fueron descritos por Engel y Arahata39 y Drachman40.
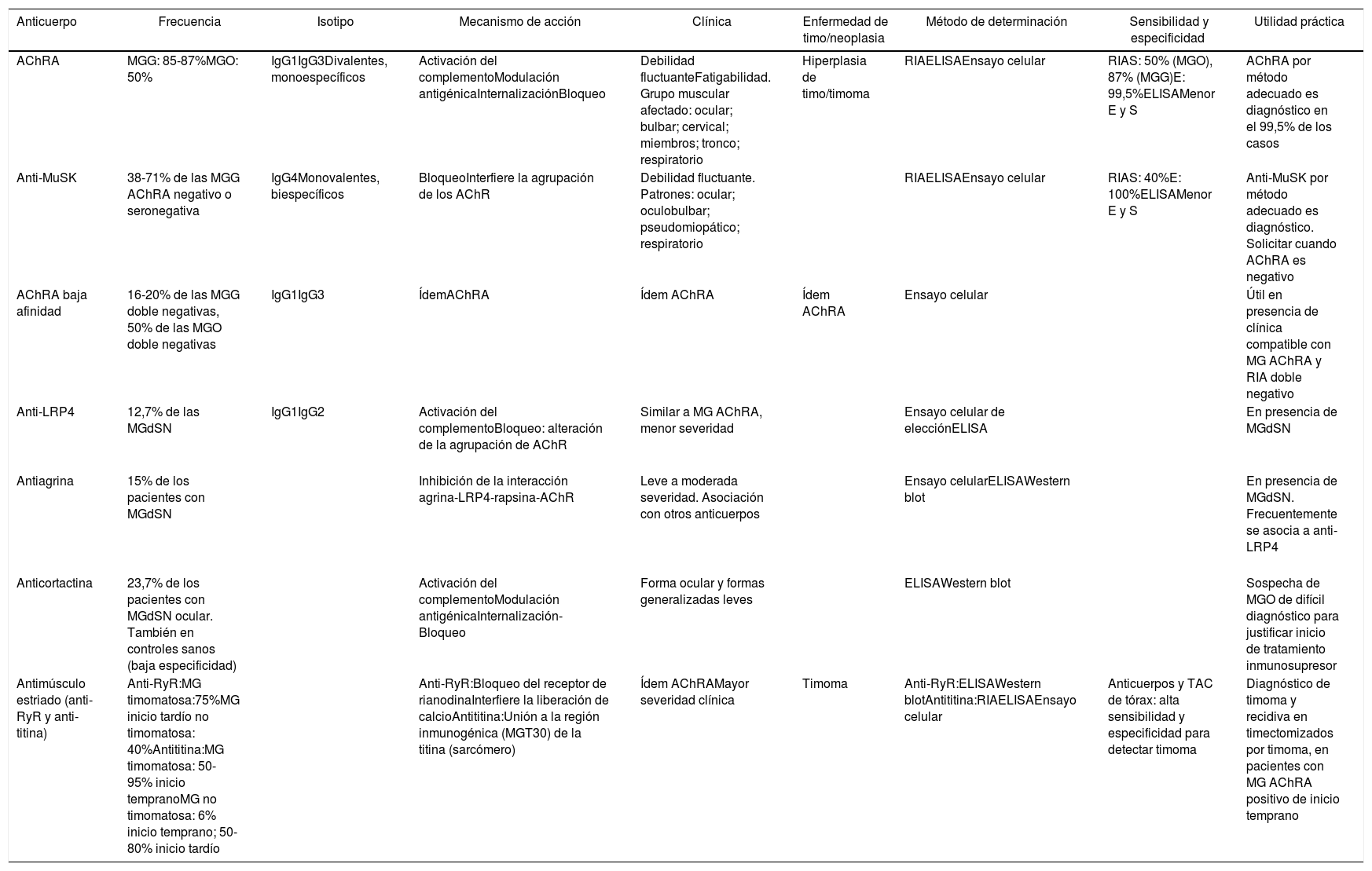
Enfermedad postsináptica: miastenia gravis
| Anticuerpo | Frecuencia | Isotipo | Mecanismo de acción | Clínica | Enfermedad de timo/neoplasia | Método de determinación | Sensibilidad y especificidad | Utilidad práctica |
|---|---|---|---|---|---|---|---|---|
| AChRA | MGG: 85-87%MGO: 50% | IgG1IgG3Divalentes, monoespecíficos | Activación del complementoModulación antigénicaInternalizaciónBloqueo | Debilidad fluctuanteFatigabilidad. Grupo muscular afectado: ocular; bulbar; cervical; miembros; tronco; respiratorio | Hiperplasia de timo/timoma | RIAELISAEnsayo celular | RIAS: 50% (MGO), 87% (MGG)E: 99,5%ELISAMenor E y S | AChRA por método adecuado es diagnóstico en el 99,5% de los casos |
| Anti-MuSK | 38-71% de las MGG AChRA negativo o seronegativa | IgG4Monovalentes, biespecíficos | BloqueoInterfiere la agrupación de los AChR | Debilidad fluctuante. Patrones: ocular; oculobulbar; pseudomiopático; respiratorio | RIAELISAEnsayo celular | RIAS: 40%E: 100%ELISAMenor E y S | Anti-MuSK por método adecuado es diagnóstico. Solicitar cuando AChRA es negativo | |
| AChRA baja afinidad | 16-20% de las MGG doble negativas, 50% de las MGO doble negativas | IgG1IgG3 | ÍdemAChRA | Ídem AChRA | Ídem AChRA | Ensayo celular | Útil en presencia de clínica compatible con MG AChRA y RIA doble negativo | |
| Anti-LRP4 | 12,7% de las MGdSN | IgG1IgG2 | Activación del complementoBloqueo: alteración de la agrupación de AChR | Similar a MG AChRA, menor severidad | Ensayo celular de elecciónELISA | En presencia de MGdSN | ||
| Antiagrina | 15% de los pacientes con MGdSN | Inhibición de la interacción agrina-LRP4-rapsina-AChR | Leve a moderada severidad. Asociación con otros anticuerpos | Ensayo celularELISAWestern blot | En presencia de MGdSN. Frecuentemente se asocia a anti-LRP4 | |||
| Anticortactina | 23,7% de los pacientes con MGdSN ocular. También en controles sanos (baja especificidad) | Activación del complementoModulación antigénicaInternalización-Bloqueo | Forma ocular y formas generalizadas leves | ELISAWestern blot | Sospecha de MGO de difícil diagnóstico para justificar inicio de tratamiento inmunosupresor | |||
| Antimúsculo estriado (anti-RyR y anti-titina) | Anti-RyR:MG timomatosa:75%MG inicio tardío no timomatosa: 40%Antititina:MG timomatosa: 50-95% inicio tempranoMG no timomatosa: 6% inicio temprano; 50-80% inicio tardío | Anti-RyR:Bloqueo del receptor de rianodinaInterfiere la liberación de calcioAntititina:Unión a la región inmunogénica (MGT30) de la titina (sarcómero) | Ídem AChRAMayor severidad clínica | Timoma | Anti-RyR:ELISAWestern blotAntititina:RIAELISAEnsayo celular | Anticuerpos y TAC de tórax: alta sensibilidad y especificidad para detectar timoma | Diagnóstico de timoma y recidiva en timectomizados por timoma, en pacientes con MG AChRA positivo de inicio temprano |
AChR: receptor de acetilcolina; anti-RyR: anticuerpo contra el receptor de rianodina; CPCP: carcinoma de pulmón de células pequeñas; E: especificidad; IT: inmunotransferencia; LRP4: proteína 4 asociada con el receptor de lipoproteínas de baja densidad; MG: miastenia gravis; MGG: miastenia gravis generalizada; MGdSN: miastenia gravis doble seronegativa; MGO: miastenia gravis ocular; RIA: radioinmunoanálisis; S: sensibilidad; TAC: tomografía axial computarizada.
En el 85% de los pacientes con fenotipo generalizado (MGG) se detectan AChRA41. En el 38-71% de los pacientes con MGG AChRA negativo se detectan anticuerpos anti-MuSK42–45. En algunos individuos con MG doble seronegativa (MGdSN), AChRA y anti-MuSK negativos, se han identificado anticuerpos contra la proteína LRP446,47, antirreceptor de acetilcolina de baja afinidad48 y antiagrina, entre otros49. Otros anticuerpos contra antígenos extracelulares e intracelulares tienen un rol menos claro en la patogenia de la miastenia y podrían ser biomarcadores50. Además de la confirmación del diagnóstico, la identificación de autoanticuerpos es importante para la estratificación en subgrupos de pacientes con MG, los que pueden diferir en sus manifestaciones, pronóstico y terapéutica.
Anticuerpo antirreceptor de acetilcolinaClínicaLa característica particular de la MG es la debilidad fluctuante, que empeora durante el día y con la actividad física y mejora con el reposo. La MG ocular es la forma de presentación más frecuente, limitada a los músculos oculomotores y elevador del párpado, con diplopía y ptosis51. La MGG se extiende a otros grupos musculares y ocurre dentro de los 2 años subsiguientes en el 80% de los casos52,53. Si comienza antes de los 40 años, el predominio es femenino 3:1 y el timo generalmente es hiperplásico, mientras que en la MG de inicio tardío la relación mujer: hombre es 3:2, el título de AChRA suele ser menor que en el grupo de inicio temprano y el timo está normal o atrófico54. El 10-15% de los pacientes tienen un timoma, más prevalente en mayores de 50 años55. El 40-50% de los timomas (incidencia 0,13/100.000/año) se asocian a MG55. La crisis miasténica ocurre en el 20% de los pacientes con MG y la remisión completa en el 10-20% de los casos 56. La MG timomatosa se considera una enfermedad más grave y su pronóstico depende principalmente de la terapia inmunosupresora prolongada. El timoma se considera un factor pronóstico negativo debido a una mayor gravedad de los síntomas y a una capacidad de respuesta reducida a los tratamientos de primera línea56.
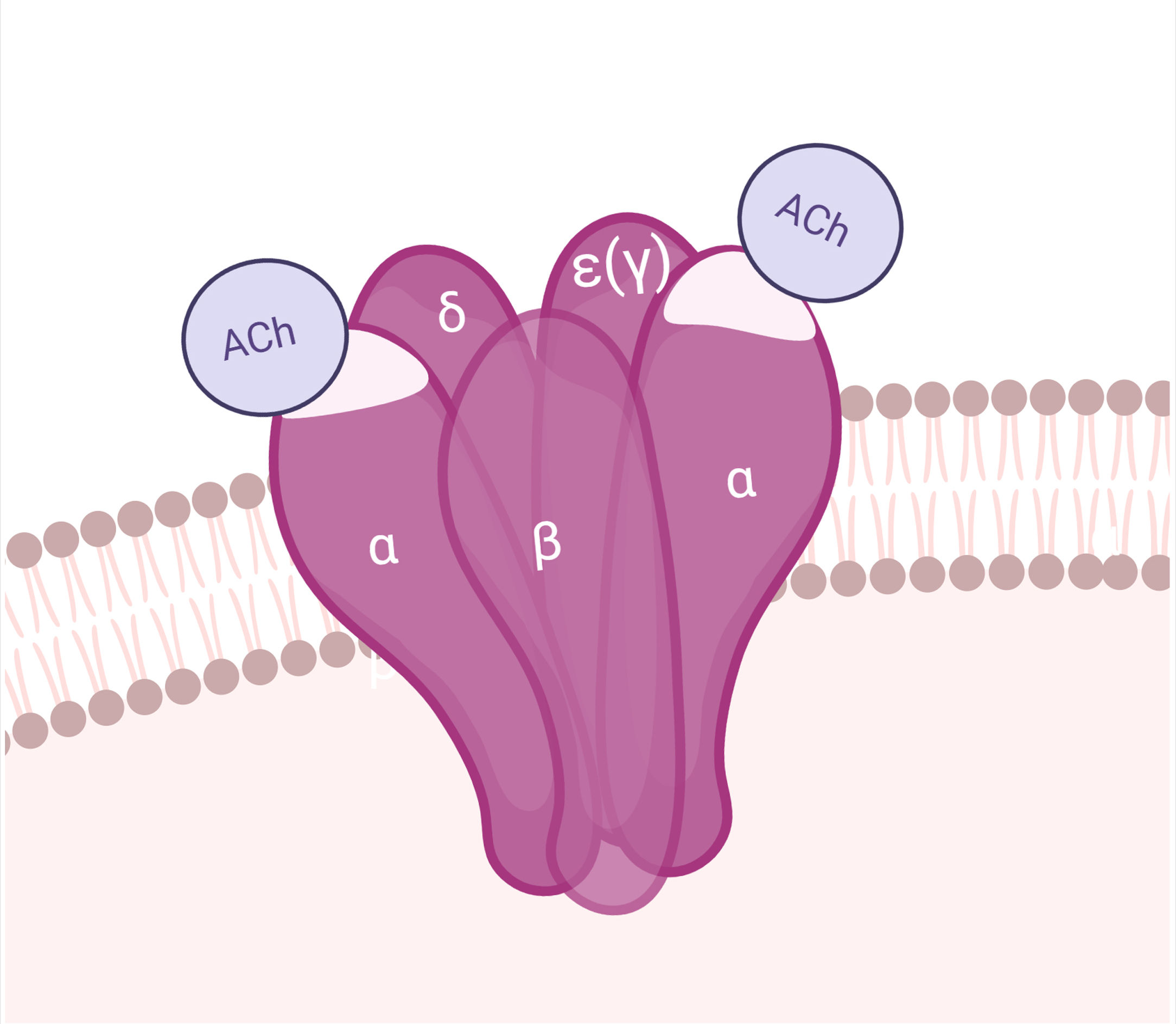
AntígenoEl AChR es una macromolécula pentamérica transmembrana, compuesta por sendas subunidades β, δ y ɛ, y 2 subunidades α en la UNM del adulto (la subunidad ɛ reemplaza a la γ de la placa fetal). Las subunidades del AChR se organizan alrededor de un canal central permeable a cationes. Cada AChR tiene 2 sitios de unión a ACh, dispuestos entre las subunidades α y δ, y α y ɛ. En la subunidad α se encuentra la región inmunogénica principal (MIR)29. La apertura del canal iónico permite el influjo de sodio y la despolarización del sarcolema (figs. 1 y 4).
Anticuerpos
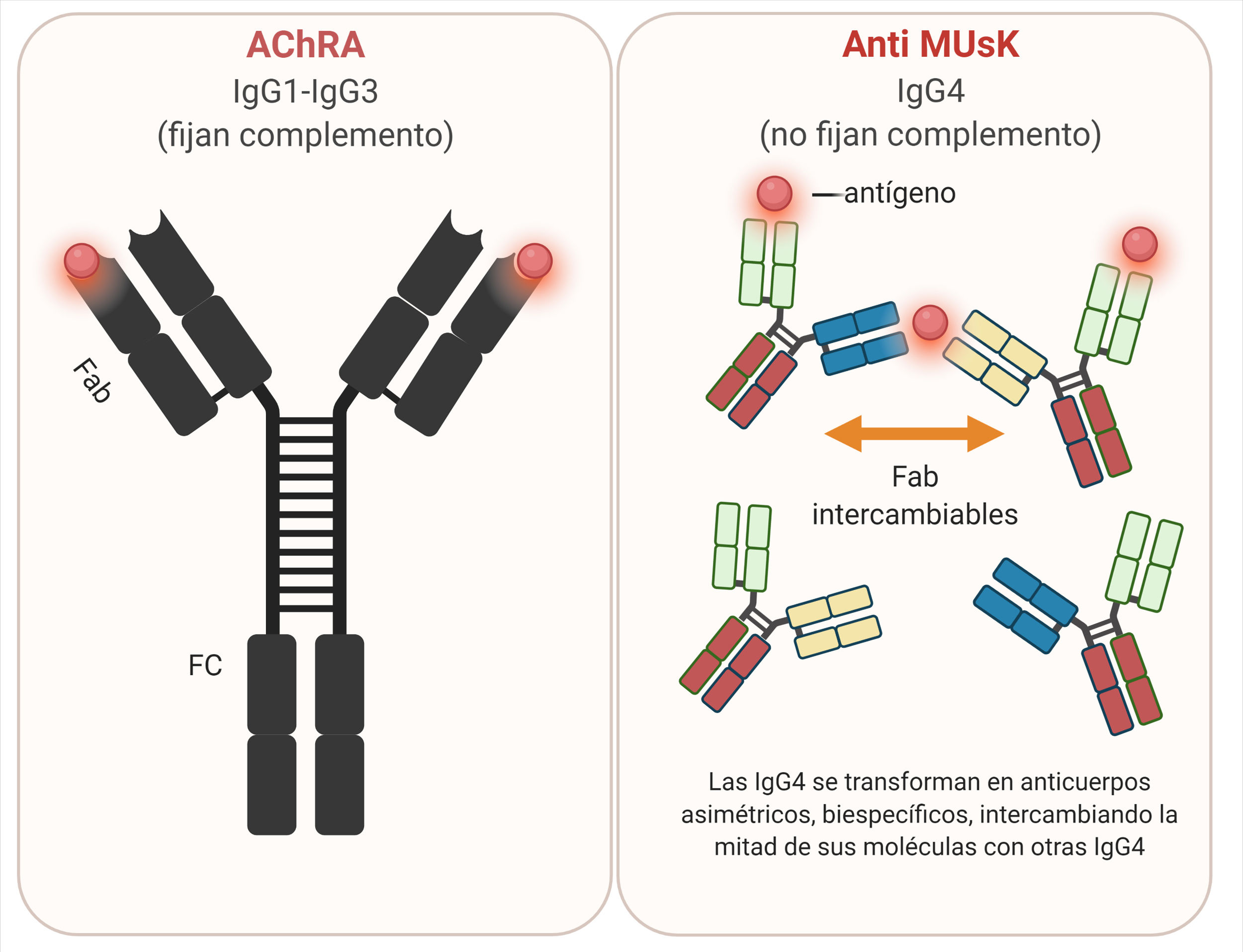
Los AChRA son predominantemente IgG1 e IgG3, divalentes, monoespecíficos, que se unen mayormente a la MIR del AChR y tienen capacidad de activar el complemento57.
Mecanismo patogénico:
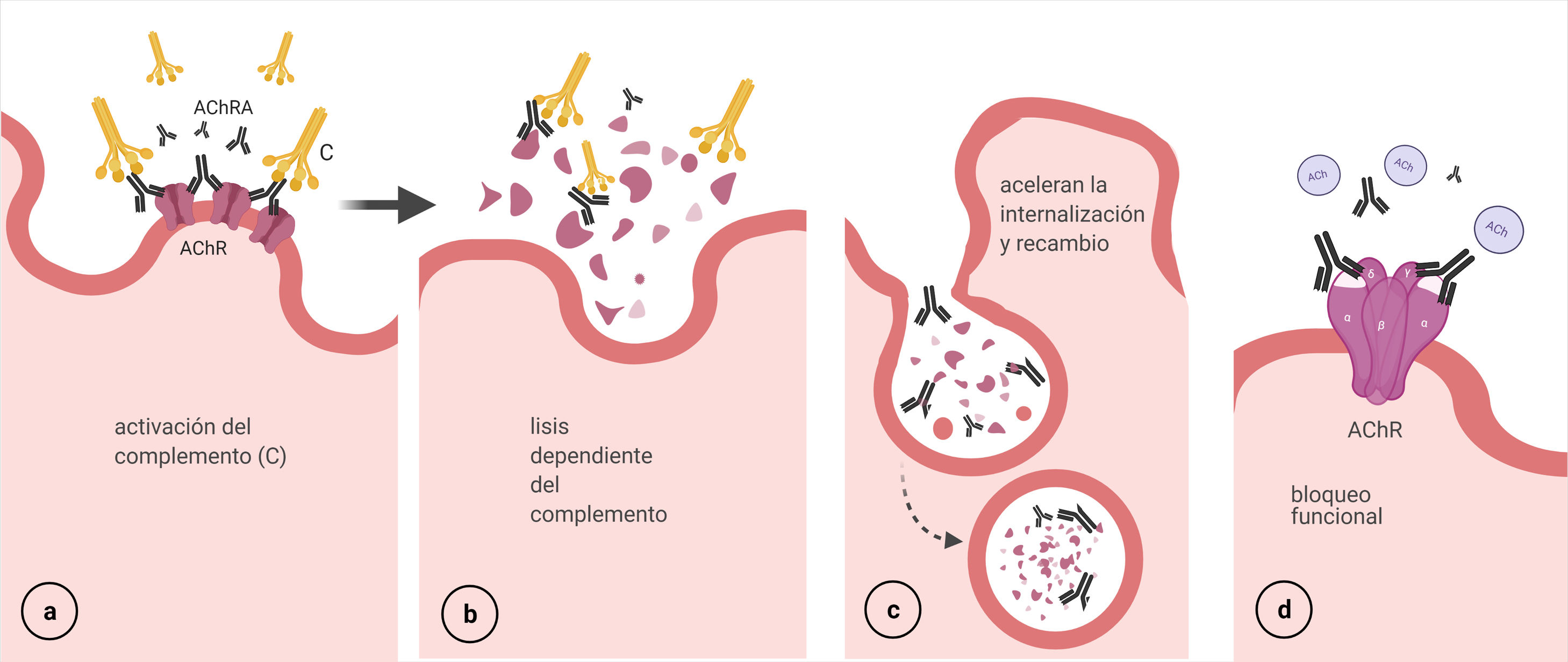
- a)
Activación del complemento: AChRA IgG1 e IgG3 son los más eficientes para la fijación del complemento. Se activa la vía clásica a partir de la unión del componente C1q al complejo AChRA-receptor, que finaliza con la formación del complejo de ataque en la membrana postsináptica y consecuentemente su destrucción57 (fig. 5 a y b).
- b)
Modulación antigénica: los AChRA (IgG) tienen funcionalidad divalente, es decir, pueden unirse a 2 moléculas del AChR diferentes, lo que provoca una internalización acelerada y la degradación de los receptores58 (fig. 5 c).
- c)
Bloqueo funcional: los anticuerpos se unen a la subunidad α (MIR) e interfieren con la unión de las moléculas de ACh40 (fig. 5 d).

El daño de la membrana postsináptica parece ser el determinante de la disfunción de la UNM59.
FrecuenciaLos AChRA están presentes en el 50% de los pacientes con MG ocular y en el 85% de las formas generalizadas41.
Métodos de determinaciónRIA es el método recomendado. ELISA es un método sensible pero las reacciones positivas débiles requieren confirmación por RIA. El ensayo celular detecta anticuerpos dirigidos contra AChR «agrupados» o AChRA de baja afinidad.
Sensibilidad y especificidadLa determinación de AChRA por RIA posee una sensibilidad del 87% para MGG y del 50% para MG ocular, y una especificidad del 99,5%60. La correspondencia entre los niveles de AChRA por ELISA y RIA usando AChR humanos es r=0,9661.
Valores de referenciaSe considera título positivo de AChRA un valor mayor de 0,5nM/L, intermedio entre 0,2-0,5 y negativo menor de 0,2nM/L41. Falsos positivos: gemelos monocigotos, timoma, discinesia tardía, pacientes con artritis reumatoidea con penicilamina, pacientes con títulos antitiroideos incrementados, hepatitis, lupus, enfermedad injerto contra huésped y neuromielitis óptica62. Es posible detectar la presencia de AChRA en pacientes con timoma sin MG, sin embargo, títulos mayores de 0,3nM/L previos a la timectomía se asocian con el desarrollo de la enfermedad poscirugía en el 7,6% de los casos63.
Utilidad en la prácticaEs la primera determinación serológica a solicitar. Un AChRA positivo por RIA es diagnóstico de enfermedad en el 99,5% de los casos. La correlación entre la concentración de AChRA y la gravedad de la enfermedad es controvertida, alguna evidencia demuestra que tal relación surge cuando se compara con el título de los anticuerpos dirigidos a MIR, subclase IgG164. Los pacientes con timoma tienen AChRA en casi todos los casos56.
Anticuerpo antitirosina cinasa específica de músculoClínicaSe han descrito 4 fenotipos asociados con anti-MuSK: oculobulbar, respiratorio, pseudomiopático65 y restringido a músculos extraoculares66. La afección del centro respiratorio en estos pacientes ha sido demostrada (De Vito y Mazia, en publicación) y estaría en relación con la mayor frecuencia de crisis respiratorias44,45. Se suele observar sarcopenia y atrofia lingual13. Otras características son la ausencia de enfermedad del timo y la respuesta especialmente satisfactoria a plasmaféresis65 y a rituximab67. Los inhibidores de la acetilcolinesterasa suelen ser menos efectivos o hasta contraproducentes45.
AntígenoMuSK es una proteína de membrana con 3 dominios similares a inmunoglobulinas (Ig-like) en la región extracelular, en donde se une el ligando68. Es activada por la agrina de la motoneurona, la cual se une a LRP4; esta aumenta la dimerización de MuSK, dispara la activación de la cinasa, selecciona Dok7 e induce una cascada de señalización que dirige la diferenciación possináptica42,69. Además, MuSK ayuda a anclar la acetilcolinesterasa mediante una hélice de colágeno Q (ColQ)69 (fig. 1).
AnticuerposLos anticuerpos anti-MuSK son mayormente IgG4, con una baja proporción de IgG170. La IgG4 no activa la cascada del complemento y es escasamente eficiente para reaccionar de forma cruzada con antígenos idénticos, debido a un proceso conocido como intercambio del brazo Fab, lo que resulta en anticuerpos monovalentes y biespecíficos71,72 (fig. 6).
Mecanismo patogénico
Bloqueo funcional: anti-MuSK inhibe la interacción entre MuSK-LRP4 a través de la unión al primer dominio Ig-like de MuSK, alterando la agrupación de los AChR. También interfiere la unión MuSK-ColQ, provocando una reducción de la concentración de acetilcolinesterasa en la hendidura sináptica72.
FrecuenciaLos pacientes con anti-MuSK representan el 38-71% de los pacientes con MGG AChRA negativo29. Ensayos celulares demostraron la presencia de anti-MuSK en el 8-13% de los sueros doble seronegativos73. La coexistencia de los anticuerpos anti-MuSK y AChRA es rara74; en Argentina, la frecuencia es del 15% (Mazia et al., datos en publicación).
Métodos de determinaciónRIA, ELISA, ensayo celular. RIA es el método recomendado42,43.
Sensibilidad y especificidadRIA: sensibilidad del 40% y especificidad del 100% en MGG AChRA negativo70. ELISA con MuSK recombinante en células transfectadas: sensibilidad del 70% y especificidad de 100% en MGG AChRA negativo42.
Valores de referenciaSe considera anti-MuSK positivo valores>0,05nM/L.
Utilidad en la prácticaSe debe considerar en pacientes con MG-AChRA negativo. La presencia de anti-MuSK con clínica compatible indica enfermedad. Los títulos de anti-MuSK IgG4 se correlacionan con la severidad de la enfermedad, y se reducen con los tratamientos inmunosupresores75.
Anticuerpos dirigidos contra el receptor de acetilcolina de baja afinidad o anticuerpos contra receptores de acetilcolina agrupadosAlgunos pacientes con hallazgo negativo de AChRA por RIA tienen un cuadro clínico indistinguible de la MG-AChRA positivo; esto apuntó a la posible existencia de anticuerpos incapaces de unirse a los AChR en solución, pero capaces de unirse a los AChR densamente agrupados, como sucede in vivo, en donde pueden unirse divalentemente con receptores adyacentes. Estos anticuerpos se conocen como AChRA de baja afinidad57; son IgG1 predominantemente. Provocan bloqueo funcional, activación de complemento y modulación antigénica. La frecuencia es de 16-20% en MGG doble seronegativa y de 50% en MG ocular AChRA negativo29. La clínica es similar a la MG-AChRA positivo, e incluso puede hallarse enfermedad del timo76. Para la determinación de estos anticuerpos se utilizan células HEK transfectadas con ADN de subunidades de AChR y rapsina.
Anticuerpo antiproteína 4 asociada con el receptor de lipoproteínas de baja densidadClínicaLa clínica de MG anti-LRP4 se asemeja a la MG-AChRA, con manifestaciones leves o moderadas y similar respuesta terapéutica46. Una mayor gravedad se observa en los pacientes con combinación de anticuerpos46,47. En los pacientes con MGdSN, anti-LRP4 se combina frecuentemente con antiagrina y el inicio generalizado es más común49.
AntígenoLa LRP4 es una proteína transmembrana cuyo dominio extracelular se une a agrina y MuSK56. Es fundamental para la activación de MuSK, el agrupamiento de AChR y la correcta conformación de la UNM29 (fig. 1).
AnticuerposLos anti-LRP4 son IgG1 e IgG246,47.
Mecanismo patogénicoBloqueo funcional de la interacción agrina-LRP4-MuSK. Activación del complemento47.
FrecuenciaCorresponden al 12,7% de las MGdSN49. Se observa variabilidad entre los diferentes métodos y países, y asociación de anti-LRP4 con AChRA, anti-MuSK46 y más comúnmente con antiagrina49.
Métodos de determinaciónValores de referenciaTítulo positivo>0,019nM47,49. Anti-LRP4 y antiagrina se consideran positivos con títulos>2,5DE de los controles49.
Utilidad en la prácticaSe solicita la determinación de anti-LRP4 en presencia de MGdSN.
Anticuerpos antimúsculo estriado (antititina, antirreceptor de rianodina y otros)Anticuerpos que aparentemente no contribuyen a la patogenia de la MG, dirigidos contra proteínas intracelulares del músculo estriado, tales como antititina, antirreceptor de rianodina (RyR), antiactina, antimiosina, antitropomiosina, entre otros, pueden detectarse en el suero de pacientes con MG-AChRA positivo43.
ClínicaLa presencia de anti-RyR y antititina ha sido correlacionada con una mayor severidad de la enfermedad77.
AntígenosRyR es un canal que libera calcio desde el retículo sarcoplásmico, localizado en la unión con el túbulo T del sarcolema. Hay 3 tipos: RyR1 (músculo esquelético), RyR2 (músculo cardíaco) y RyR3 (cerebro)77. Titina es una proteína gigante del miocito (fig. 1) que se extiende a lo largo de todo el sarcómero y provee estabilidad y resistencia pasiva al estiramiento78.
AnticuerposLos anticuerpos dirigidos contra las proteínas miofibrilares tienen reacción cruzada con células mioides del timo. Se los encuentra en el 80% de los pacientes miasténicos con timoma79. La presencia de estos anticuerpos ha sido demostrada en biopsia de músculo de paciente con MG y podrían predecir un severo defecto sobre el desarrollo y la función del músculo80.
Mecanismo patogénicoAnti-RyR: inhibe la liberación de calcio uniéndose a la región inmunogénica principal del RyR. Hay anti-RyR que inhiben la interacción entre RyR y dihidropiridina, y en estos casos la severidad de la MG sería mayor78.
Antititina: se une a la región inmunogénica principal de la titina, MGT30, próxima a la unión de la banda A y la banda I, y altera el sarcómero. Otro mecanismo es a través de una respuesta inmune celular78.
FrecuenciaAnti-RyR: baja frecuencia en la MG de inicio temprano, 40% en la MG de inicio tardío y 75% en la MG con timoma.
Antititina: en el 20-40% de las MG-AChRA positivo. En la MG no timomatosa, en el 6% cuando el inicio es temprano y en el 50-80% cuando el inicio es tardío. En la MG timomatosa de inicio temprano se detecta en el 50-95%29. La ausencia de AChRA aleja la posibilidad de timoma79.
Método de detecciónAnti-RyR: ELISA, Western blot.
Anti-titina: ELISA, RIPA MGT30 con 125I, ensayo celular43,79.
Sensibilidad y especificidadLa combinación de anticuerpos y tomografía de tórax tiene una alta especificidad y sensibilidad para detectar timoma55.
Utilidad prácticaLa determinación de anticuerpos antititina y anti-RyR es útil para diagnosticar timoma en pacientes con MG-AChRA positivo de inicio temprano y recidiva en timectomizados. Estos anticuerpos también se asocian con una mayor severidad de la enfermedad.
Anticuerpos contra otras proteínasOtros anticuerpos cuya patogenicidad, especificidad y valor diagnóstico o pronóstico no han sido completamente caracterizados están dirigidos contra las proteínas agrina29,49, cortactina50, rapsina29, acetilcolinesterasa, ColQ, colágeno xiii y Kv1.4 presináptico29 (fig. 1). Las principales características se describen en la tabla 2.
ConclusionesEn la unión neuromuscular, la transmisión de señales depende de canales y complejos proteicos, pre y postsinápticos. La autoinmunidad dirigida a estos blancos antigénicos puede ser primaria o paraneoplásica; los tumores de timo y pulmón deben ser particularmente considerados.
El RIA es el método usualmente recomendado para la detección de la mayoría de los anticuerpos. Los métodos basados en ensayo celular son realizados en laboratorios especiales y son requeridos para la determinación de anticuerpos particulares, como los AChRA de baja afinidad.
En todos los casos, la sensibilidad y la especificidad del anticuerpo dependen del método utilizado para su medición. Diversos anticuerpos, tales como los dirigidos a estructuras presinápticas (VGCC y complejo VGKC), poseen una sensibilidad y especificidad en relación con el título sérico.
Algunos de los anticuerpos dirigidos al complejo VGKC se ligan al canal de potasio propiamente dicho, pero, mayormente, lo hacen a las otras proteínas (CASPR2 y LGI1), y esto se traduce en diferentes fenotipos.
Los anticuerpos dirigidos a la postsinapsis son los más frecuentes en la enfermedad autoinmune de la neurotransmisión, específicamente el AChRA, cuyo mecanismo patogénico es el más conocido. La positividad del AChRA o del anti-MuSK mediante RIA, independientemente del título, indica con elevada certeza el diagnóstico de MG. Se ha demostrado correlación entre la concentración sérica de anti-MuSK y la severidad de la enfermedad. En el caso de los AChRA, no existe un criterio unánime sobre esta correlación.
La ausencia de anticuerpos o un título indeterminado obliga a repetir la serología si la sospecha clínica persiste. La positividad puede aparecer y el título puede cambiar durante la evolución de la enfermedad. La reiteración del RIA es igualmente recomendada en presencia de un timoma con un resultado de AChRA negativo.
El hallazgo de anticuerpos dirigidos a antígenos intracelulares del músculo estriado (titina y RyR) con un AChRA positivo soportan fuertemente el diagnóstico de timoma, más aún si se combinan con la existencia de una imagen anormal en el mediastino. La MG asociada a timoma tiene peor pronóstico.
La utilidad de un anticuerpo como marcador específico de enfermedad requiere criterios estrictos. El conocimiento de la clínica y la electrofisiología es relevante, fundamentalmente cuando la sensibilidad y la especificidad de los anticuerpos son bajas. La disautonomía indica afección presináptica. Mioquimias, fasciculaciones, hiperexcitabilidad y síntomas centrales evidencian anticuerpos contra el complejo VGKC. Los signos de impregnación orientan hacia un cuadro paraneoplásico.
Finalmente, se debe jerarquizar la precocidad con que se hace el diagnóstico, dado que se trata de enfermedades potencialmente tratables y pasibles de remisión.
Conflicto de interesesEl grupo de trabajo no presenta ningún conflicto de intereses en relación con la redacción de este artículo.
Agradecemos las ilustraciones a Mariana Bendersky (biorender.com), del Servicio de Neurología Infantil, Hospital Italiano de Buenos Aires, Instituto Argentino de Investigaciones Neurológicas (IADIN), Facultad de Medicina, Universidad de Buenos Aires, EnyS-CONICET.
