




El síndrome del cromosoma 20 en anillo se caracteriza por epilepsia refractaria, estatus epiléptico no convulsivo, electroencefalograma característico, trastorno de conducta y discapacidad intelectual leve-moderada.
Caso clínicoVarón de 6 años con discapacidad intelectual leve con movimientos involuntarios en vigilia de flexoextensión de dedos de manos y muñecas, asociando dudosa desconexión del medio intermitente. En videoelectroencefalograma se confirma estatus epiléptico no convulsivo. En cariotipo se identifica r(20) de novo.
ConclusiónDebemos sospechar r(20) en todo paciente con epilepsia con anomalías frontales, estatus epiléptico no convulsivo y discapacidad intelectual, aunque presente fenotipo normal, y comenzar el estudio por el cariotipo.
Ring chromosome 20 syndrome is characterized by refractory epilepsy, non-convulsive epilepticus status, characteristic changes on electroencephalogram, behavioral disorder and mild to moderate intellectual disability.
Clinical caseSix-year-old man presented with mild intellectual disability and involuntary waking flexion-extension movements of the fingers and wrists, associating intermittent episodes of staring spells. EEG showed non-convulsive status epilepticus. karyotype analysis identified r(20) de novo.
ConclusionWe should suspect r(20) in all patients with frontal lobe epilepsy, non-convulsive epilepticus status and intellectual disability, even if the patient has a normal phenotype, and we should include karyotype as part of the initial evaluation.
El síndrome del cromosoma 20 en anillo –r(20)– es una enfermedad rara de base genética conocida. Resulta de roturas en los extremos de ambos brazos de dicho cromosoma, con posterior fusión de las regiones rotas, produciéndose un anillo continuo1.
Desde su descripción por Atkins en 19722 se han documentado poco más de 150 casos en la literatura, aunque la prevalencia exacta es desconocida3. En la mayoría de los casos se trata de una condición espontánea4 sin riesgo de recurrencia, aunque se han descrito de forma excepcional algunas familias en las que se ha visto transmisión de padres a hijos5.
Se caracteriza por presentar epilepsia refractaria con crisis frontales, estatus epiléptico no convulsivo (SENC), electroencefalograma (EEG) característico, trastorno de conducta y discapacidad intelectual (DI) leve-moderada, sin alteraciones fenotípicas reseñables. La mayoría de los pacientes suelen presentar un cociente intelectual normal o límite hasta el comienzo de la epilepsia. Esta suele ser el primer síntoma del síndrome, y tras su aparición, más del 70% de los pacientes presentan deterioro cognitivo. Parece existir una correlación entre el porcentaje de células afectadas, el cociente intelectual y la edad de inicio de las crisis. Mayor grado de mosaicismo en las células con r(20) se asocia con un comienzo más temprano de crisis, resistencia a fármacos antiepilépticos y mayor trastorno de conducta3,6. Se postula la implicación de los genes CHRNA4 y KCNQ2 en el desarrollo de la epilepsia, ambos localizados en el brazo largo del cromosoma 20 en la región 20qter7. En este trabajo presentamos un niño de 6 años con esta patología que ingresa para estudio, inicialmente por episodios paroxísticos de ambas manos, que recuerdan a estereotipias manuales.
Caso clínicoVarón de 6 años que acude a urgencias por episodios paroxísticos de 30 min de duración que inició la semana previa de forma intermitente, en vigilia. Consiste en movimientos involuntarios de flexoextensión de dedos de ambas manos y ocasionalmente muñeca, varios por segundo, rítmicos, de predominio derecho. En ocasiones asocia mirada fija con dudosa desconexión del medio durante el episodio.
Sin antecedentes familiares de interés. Hijo único de padres no consanguíneos.
Como antecedentes personales presenta seguimiento en consulta de neurología infantil el último año por discapacidad intelectual ligera con desfase curricular significativo. Embarazo y parto normales. El desarrollo psicomotor había sido normal con marcha autónoma a los 15 meses. Exploración física, neurológica, fenotipo y somatometría normales.
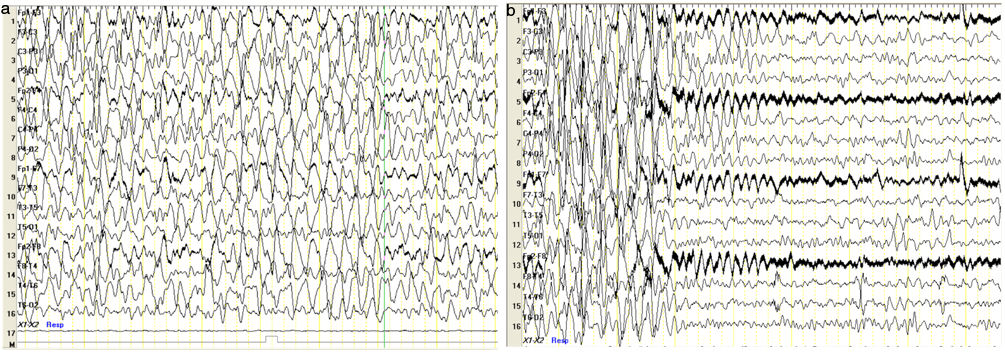
Durante uno de los episodios descritos, se objetiva en videoelectroencefalograma (vEEG) SENC (fig. 1a) que cede tras la administración de midazolam bucal (fig. 1b). En el vEEG de sueño se registran anomalías epileptiformes frontales bilaterales.
 en vigilia. Coincidiendo episodio de estatus no convulsivo (SENC) con movimientos involuntarios de flexoextensión de dedos de ambas manos y ocasionalmente muñeca, rítmicos, de predominio derecho, y desconexión intermitente del medio, se registran descargas epileptiformes generalizadas con morfología de punta onda hipervoltada a), SENC que cede tras administración de midazolam yugal b).")
Videoelectroencefalograma (vEEG) en vigilia. Coincidiendo episodio de estatus no convulsivo (SENC) con movimientos involuntarios de flexoextensión de dedos de ambas manos y ocasionalmente muñeca, rítmicos, de predominio derecho, y desconexión intermitente del medio, se registran descargas epileptiformes generalizadas con morfología de punta onda hipervoltada a), SENC que cede tras administración de midazolam yugal b).
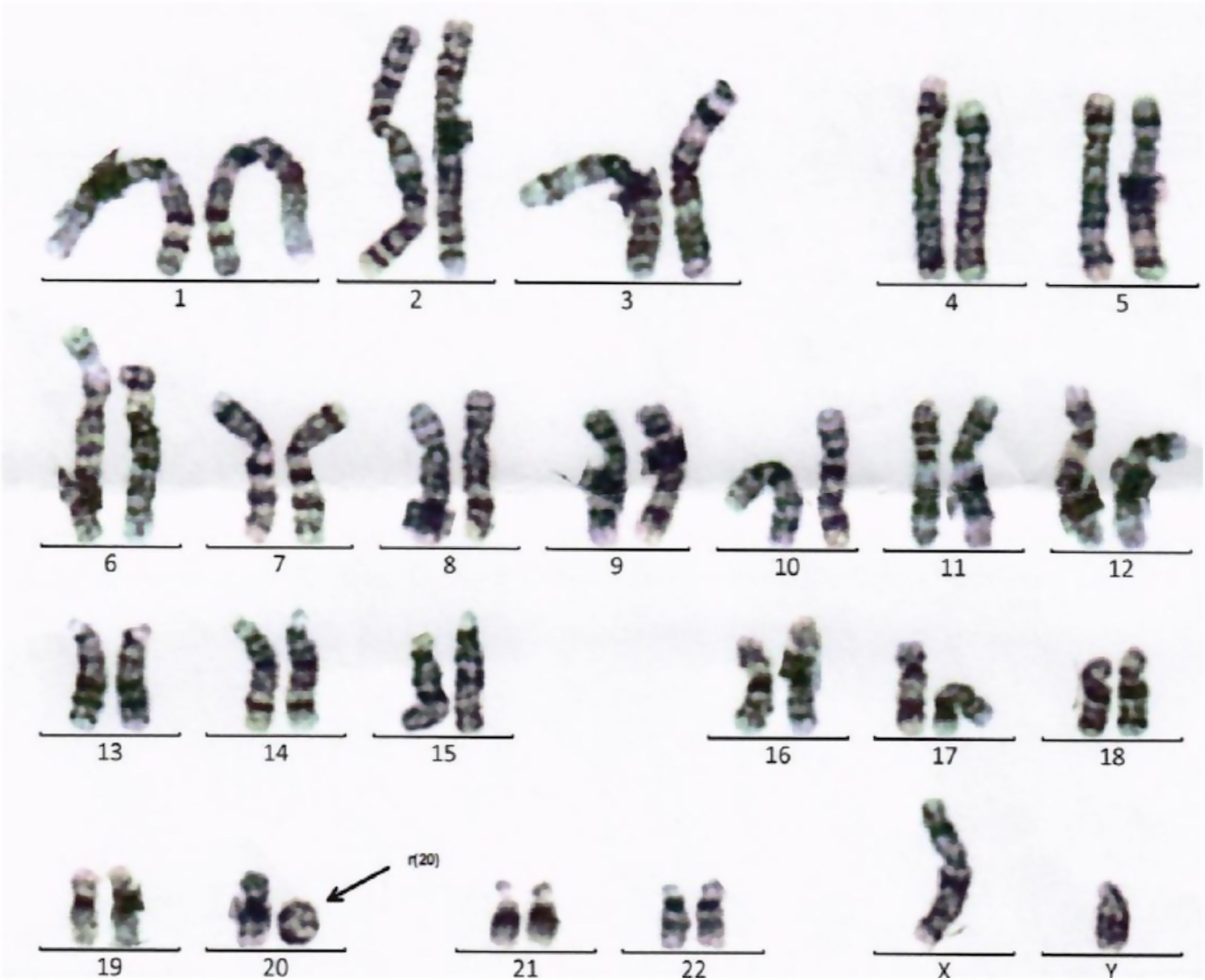
Se completa estudio con analítica sanguínea con perfil metabólico, % CDT, aminoácidos y ácidos orgánicos en sangre y orina, valoración por oftalmología con fondo de ojo y auditiva, con resultados normales. X-frágil negativo. La resonancia magnética craneal únicamente demuestra megacisterna magna en fosa posterior. En el cariotipo se identifica r(20) (fig. 2) de novo, sin mosaicismo, comprobándose pérdida de región subtelomérica del brazo corto mediante FISH. En CGH array se confirma la deleción en la citobanda 20p13 –deleción terminal 20pter que incluye los genes NRSN2 y SOX12–, candidatos para el retraso del desarrollo.

Con el diagnóstico de r(20) se inicia tratamiento con ácido valproico, precisando añadir lamotrigina cuatro meses después. Tres años más tarde se mantiene sin crisis y sin aparente deterioro cognitivo, con aceptable progresión en el aprendizaje.
ConclusionesEl cromosoma 20 en anillo es una patología muy poco frecuente (con una prevalencia menor de 1 en 1.000.0008), por ello, se debe tener un alto índice de sospecha para su diagnóstico.
Además, en ocasiones solo se puede diagnosticar mediante la realización de un cariotipo y no mediante otras pruebas genéticas como un CGH array si no existe deleción en la mutación6. Esto nos puede retrasar el diagnóstico, ya que en pocos casos de pacientes con epilepsia en el momento actual se realiza un cariotipo como parte de la batería de pruebas complementarias, solicitando inicialmente pruebas genéticas más específicas.
El EEG es característico aunque no patognomónico9. Aún así tenemos que pensar en ello en pacientes con la siguiente tríada electroclínica10,11: crisis frontales refractarias, SENC recurrente y hallazgos típicos en EEG. Respecto a las crisis frontales, se incluyen tres tipos de crisis: crisis nocturnas hiperquinéticas o hipermotoras, crisis nocturnas sutiles con mínima actividad motora solo diagnosticadas en ocasiones mediante vEEG y a veces confundidas con un arousal fisiológico9 y crisis parciales complejas, con mirada fija, confusión, con o sin automatismos orales o motores.
Respecto al SENC, para algunos autores es el patrón más característico de este síndrome12, presentándose la mayoría de los casos con una frecuencia diaria y de predominio en vigilia9.
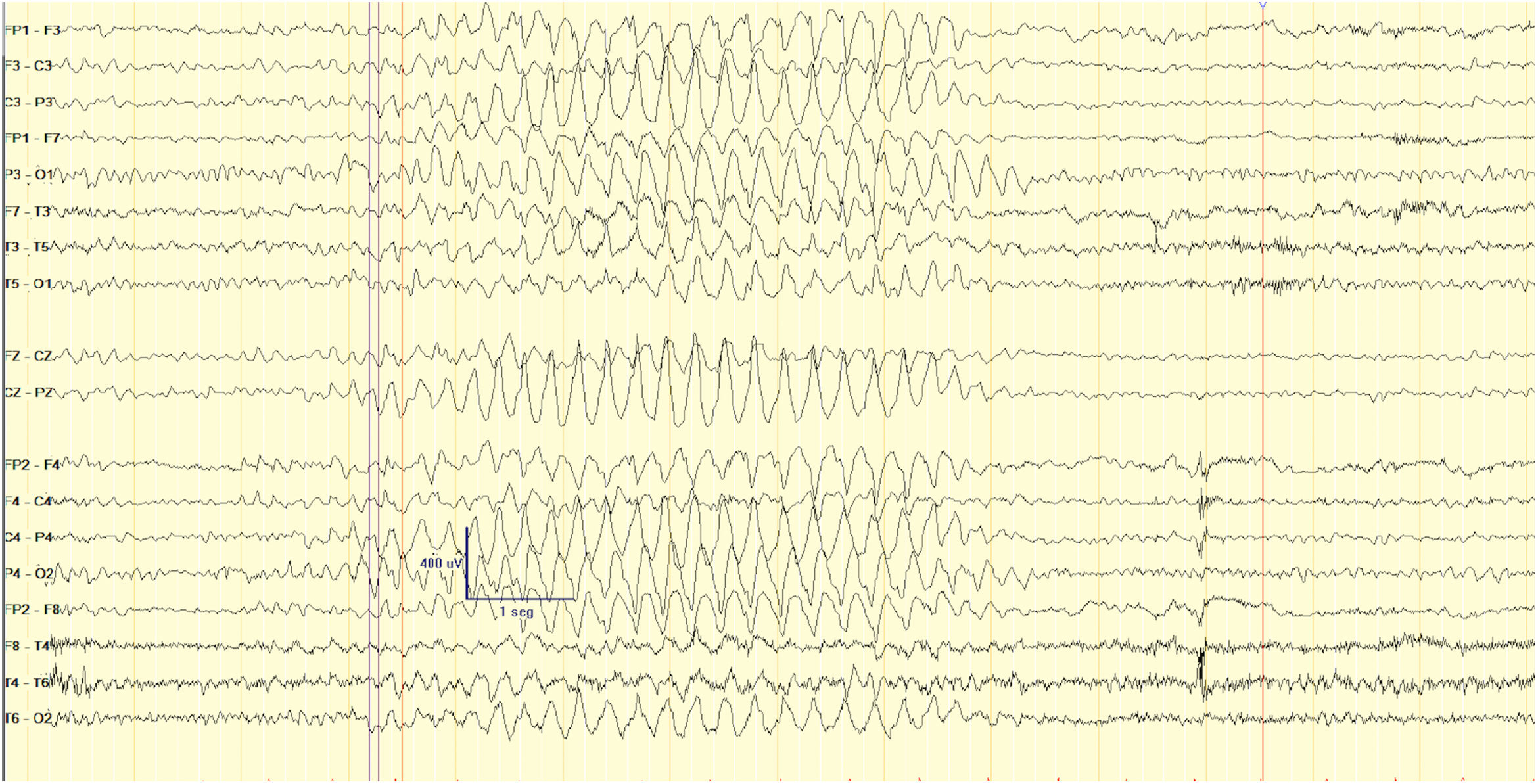
Respecto a los hallazgos típicos en EEG se describe actividad lenta hipervoltada con puntas en forma de descargas epileptiformes generalizadas (fig. 3) y de predominio frontal uni- o bilateral. Además también se observan frecuentes trenes de ondas theta en áreas frontotemporales no influenciadas por la apertura ocular, hallazgo muy específico13. Aun así también se han identificado EEG normales o con solo una lentificación difusa, sin actividad irritativa9.
.")
VEEG en vigilia. Brotes difusos de actividad delta hipervoltada, que en ocasiones se entremezclan con puntas adoptando una morfología mellada y otras se presentan con una morfología de ondas delta agudas de aspecto más triangular, de máxima amplitud variable, de breve duración (3-6 segundos).
Esta tríada electroclínica parece tener una sensibilidad (si está presente) y un valor predictivo negativo (si no lo está) cercana al 100%10. Aun así existen otras patologías que presentan dicha tríada, como son: cromosoma 10 en anillo, síndrome de Dravet, síndrome de Lennox-Gastaut, displasias corticales frontales y epilepsia con punta onda continua durante el sueño lento mal tratada con bloqueantes de canales de sodio. No obstante, no se pueden extrapolar estos hallazgos a todas las edades10, sobre todo en los menores de 1 año, aunque hay autores que comentan que el EEG interictal de estos pacientes suele aparecer a partir de los 8 años de edad14. Un EEG normal en vigilia requiere un estudio de EEG durante el sueño para un diagnóstico precoz correcto9.
Se trata de una epilepsia con frecuencia farmacorresistente no susceptible al tratamiento quirúrgico, por lo que el tratamiento es limitado9. Además suele haber un empeoramiento clínico progresivo con la edad, con un aumento de la frecuencia de crisis y mayor refractariedad al tratamiento, siendo en algún caso letal15.
El tratamiento farmacológico inicial de elección suele ser el ácido valproico, pudiendo añadirse lamotrigina o zonisamida, con mejor respuesta con la primera combinación10. También se describe la lacosamida como una buena alternativa terapéutica16 así como el estimulador del nervio vago10 para casos refractarios.
Además los pacientes que suelen tener un comportamiento y nivel cognitivo normal previo al inicio de la epilepsia, suelen empeorar de forma dramática en ambos aspectos tras su inicio17. Parece existir una relación inversamente proporcional entre la edad de inicio de las crisis y el porcentaje de mosaicismo en sangre periférica18, habiéndose descrito pacientes con un comienzo de epilepsia tardío, con un curso más suave, con remisión de las crisis en algún caso6. Según algunos autores, pacientes afectos con el 100% de células con r20 deberían considerarse como un fenotipo diferente, con un retraso psicomotor y una epilepsia más grave19.
Nuestro caso es llamativo, dado que por una parte, la alteración genética no presenta mosaicismo, por lo que según la literatura, la epilepsia tendría que haber aparecido mucho antes de los 6 años, pero no ha sido así. Además la pérdida de material genético está localizada en el brazo corto del cromosoma 20 y no en el largo, donde se encuentran los genes CHRNA4 y KCNQ2, que se han postulado hasta la fecha como posibles responsables de la epilepsia. Posiblemente, como se ha sugerido, la epilepsia puede ser debida a una regulación anormal de estos genes por los efectos de posición en el anillo, no tanto a una pérdida de los mismos6.
El diagnóstico es difícil hasta el comienzo de la epilepsia, que suele ser el primero signo.
Por tanto, en pacientes con epilepsia con crisis frontales, SENC, EEG característico, trastornos de conducta y DI sin alteraciones fenotípicas, es recomendable realizar inicialmente un cariotipo antes que otras pruebas genéticas.
Posteriormente habría que determinar los genes delecionados con CGH array. Y en caso de no deleción de 20q sospechar alteraciones génicas en CHRNA4 y KCNQ2. El conjunto de las técnicas genéticas será lo que nos dé un diagnóstico más preciso.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.
