







Las enfermedades mitocondriales comprenden un conjunto de trastornos multisistémicos que afectan al normal funcionamiento de la maquinaria energética celular. Su diagnóstico acabado requiere de una aproximación que, además de un alto índice de sospecha, involucre distintas técnicas de biología molecular y una cuidadosa selección del tejido a estudiar. Con un objetivo general de evaluación de su abordaje diagnóstico, exploramos la utilidad de un nuevo algoritmo diagnóstico y estandarizamos herramientas moleculares necesarias para su implementación.
Material y métodosSe realizó un estudio prospectivo, analítico y observacional en una cohorte de 73 pacientes con sospecha de desorden mitocondrial, asistidos en nuestro centro en el período comprendido entre mayo del 2008 y junio del 2016. Pusimos a punto desarrollos diagnósticos moleculares que incluyeron técnicas clásicas monogénicas y novedosas genómicas de alto rendimiento.
ResultadosCaracterizamos las manifestaciones neurológicas y extraneurológicas de nuestra cohorte; 61 pacientes fueron clasificados en síndromes mitocondriales clásicos, siendo LHON, MELAS y CPEO los más frecuentes. Siguiendo el algoritmo propuesto, obtuvimos un rendimiento diagnóstico molecular de un 51%, pudiendo identificar alteraciones en 37 pacientes. Mutaciones puntuales en el ADNmt fueron individualizadas en 30 pacientes, alteraciones estructurales en el genoma mitocondrial en 3 y mutaciones en genes nucleares en 4.
ConclusionesNuestros resultados confirman la utilidad del algoritmo propuesto utilizado y de las herramientas moleculares empleadas, manifestado en un alto rendimiento diagnóstico. Esto resulta de gran valor para una asistencia médica más eficiente e integral de los pacientes y familias afectados por desórdenes mitocondriales.
Mitochondrial diseases encompass a group of multi-systemic disorders affecting the normal function of the cellular energetic machine. Diagnosis requires an approach that, in addition to a high index of suspicion, involves molecular techniques and a careful selection of the tissue to be studied. With an overall objective assessment and diagnostic approach, we explored the utility of a new algorithm and standardized the molecular diagnostic tools needed for its implementation.
Material and methodsA prospective, analytical, observational study was conducted in a cohort of 73 patients with suspected mitochondrial disorder who were treated at our hospital between May 2008 and June 2016. We developed molecular diagnostic tools that included classical monogenic techniques and novel high-performance genomic techniques.
ResultsWe characterized the neurological and extra-neurological manifestations noted in our cohort. 61 patients were classified into classical mitochondrial syndromes, being LHON, MELAS and CPEO the most frequent. Following the proposed algorithm, we obtained a molecular diagnostic performance of 51%, identifying mutations in 37 patients. DNAmt mutations were identified in 30 patients. Structural rearrangements in mitochondrial genome were found in three and four in nuclear genes, respectively.
ConclusionsOur results confirm the utility of the proposed algorithm and the molecular tools used, as evidenced by a high diagnostic performance. This is of great value to a more efficient and comprehensive medical care of patients and families affected by mitochondrial disorders.
Las enfermedades mitocondriales comprenden un conjunto de trastornos multisistémicos que afectan al normal funcionamiento de la maquinaria energética celular. Dado que las proteínas pertenecientes a los procesos de la cadena respiratoria y fosforilación oxidativa (CR/OXPHOS) son codificadas por 2 genomas diferentes e independientes (ADN mitocondrial [ADNmt] y ADN nuclear [ADNn]); este grupo de enfermedades pueden deberse a mutaciones en cualquiera de ellos y seguir un modelo de herencia mitocondrial o mendeliano. Además, el ADNmt puede verse afectado por grandes rearreglos estructurales (deleciones parciales o duplicaciones), generalmente responsables de síndromes de presentación esporádica. Sumando complejidad, tanto las mutaciones puntuales del ADNmt como los grandes rearreglos suelen ser heteroplásmicos (cualidad dada por coexistencia de ADNmt salvaje y mutado en una mitocondria, célula o tejido) y cada tejido presenta un umbral de expresión clínica diferente1. Todo esto lleva a la necesidad de implementar distintas técnicas de biología molecular y una cuidadosa selección del tejido a estudiar a fin de incrementar la chance de identificar un defecto causal en las enfermedades mitocondriales1.
Con respecto a la presentación clínica, es característico su amplio pleomorfismo, pudiendo ser diagnóstico diferencial de diversos trastornos neurológicos y extraneurológicos. En consecuencia, su diagnóstico requiere de un alto índice de sospecha. Existen múltiples algoritmos propuestos, tanto a nivel local2 como a nivel internacional3-5. Sin embargo, estos deben ser actualizados teniendo en cuenta la disponibilidad de nuevas técnicas de secuenciación (como la secuenciación de alto rendimiento conocida como NGS por sus siglas en inglés) y el mayor acceso a las pruebas moleculares en general. Por otro lado, existe escasa experiencia local en esta población de pacientes en Argentina. Comprendiendo a la enfermedad mitocondrial como una patología genética, su diagnóstico no concluye hasta la detección de la mutación patogénica, lo cual excede los fines diagnósticos y la consejería genética, sino que implica el reconocimiento de posibles blancos terapéuticos objeto de ulteriores desarrollos.
En este trabajo, nuestro objetivo general es evaluar el abordaje diagnóstico de los desórdenes mitocondriales con manifestaciones en el sistema nervioso haciendo uso de una aproximación estandarizada, tanto en términos clínicos como moleculares. Para ello, como objetivos específicos nos proponemos:
- •
Explorar la utilidad de un nuevo algoritmo diagnóstico de enfermedades mitocondriales.
- •
Estandarizar herramientas de diagnóstico molecular necesarias para la implementación del mismo.
- •
Ilustrar mediante casos representativos el pleomorfismo fenotípico y la utilidad de dichas herramientas en el diagnóstico de enfermedades mitocondriales.
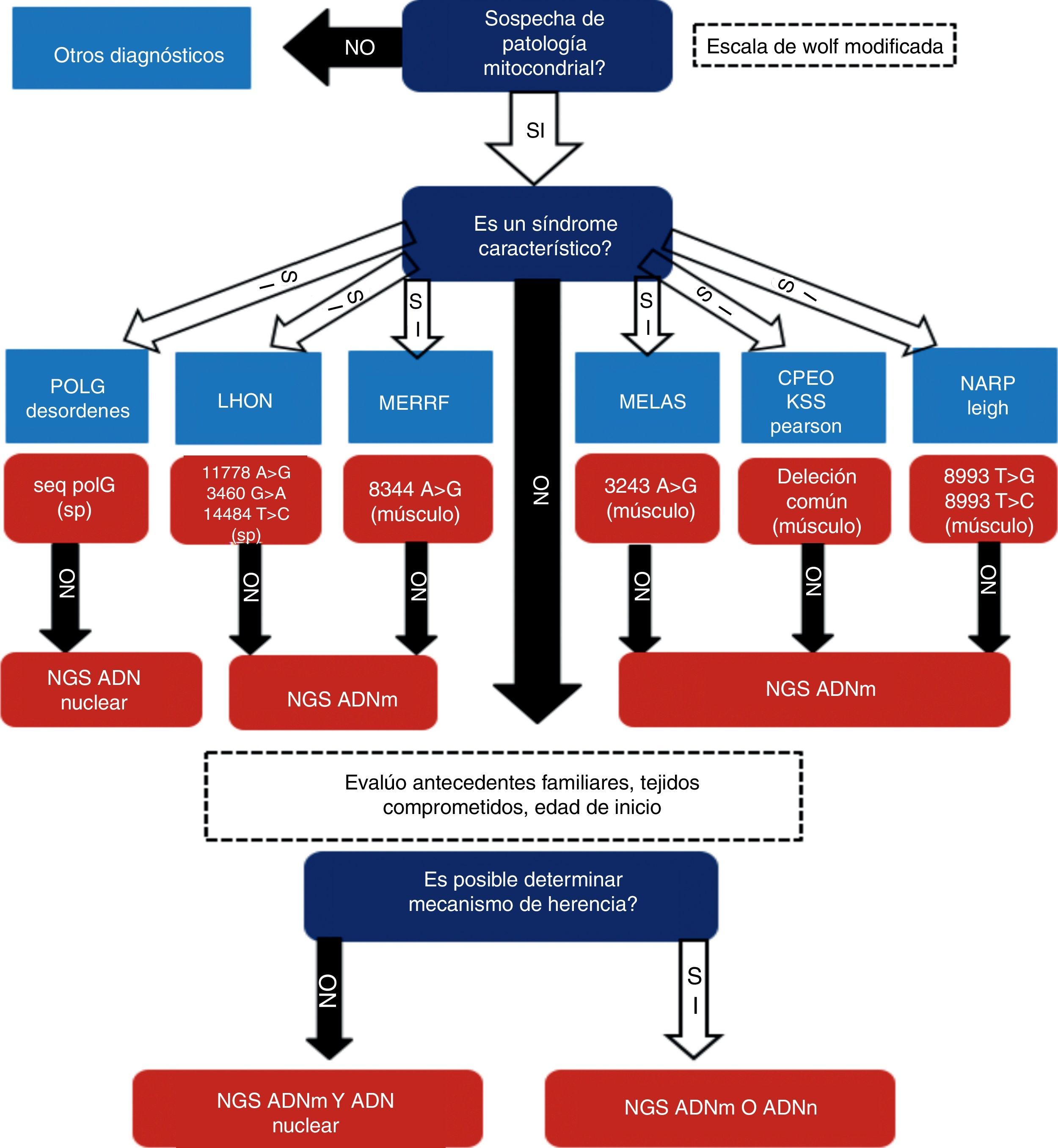
Se realizó un estudio prospectivo, analítico y observacional en una cohorte de 64 pacientes con sospecha de desorden mitocondrial asistidos en nuestro centro en el período comprendido entre mayo del 2008 y junio del 2016. Todos los pacientes brindaron consentimiento informado mediante formulario aprobado por el Comité de Ética institucional previamente a su participación en el estudio. En todos los casos se recabó información demográfica, familiar, clínica y paraclínica. En cada uno de los pacientes se siguió un enfoque diagnóstico basado en 3 etapas, modificado de una propuesta previa de uno de los autores de este trabajo2. Este algoritmo es presentado en la figura 1. Brevemente, consiste en etapas sucesivas de caracterización y direccionamiento de procedimientos diagnósticos orientados a la individualización del defecto molecular etiopatogénico. El primer paso consiste en determinar cuál es la probabilidad de que el paciente tenga una enfermedad mitocondrial. Se seleccionó la escala de Wolf6 para estratificar esta probabilidad mediante la sumatoria de signos y síntomas y de los hallazgos en pruebas bioquímicas e histopatológicas (fig. 2). En el segundo paso se determina si tiene presentación fenotípica correspondiente a un síndrome clásico o no (MELAS, MERRF, CPEO, Leigh, SANDOS, etc.). Si el paciente presenta un fenotipo clásico se realiza la búsqueda de mutaciones puntuales y/o alteraciones estructurales en el ADNmt (2-10kb) o la secuenciación de genes nucleares candidatos como C10orf2 o POLG, según corresponda. En caso de que estos estudios resulten negativos, se continúa con la secuenciación de exoma completo y/o de genoma mitocondrial, según pudiera indicar el patrón de herencia observado en la familia estudiada y evaluada en el paso 3.


Criterios de probabilidad de disfunción mitocondrial a partir de datos clínicos y de estudios complementarios. Modificada de Wolf et al.6.
Se obtuvo de cada paciente 5ml de sangre entera por venopuntura y en casos seleccionados se obtuvo tejido muscular mediante biopsia. Las muestras se conservaron codificadas a –20 °C y/o nitrógeno hasta su posterior procesamiento. Se purificó luego ADN genómico total utilizando sistemas comerciales siguiendo las instrucciones del fabricante. Este fue conservado anonimizado hasta su posterior procesamiento. A partir de la direccionalidad propuesta por el algoritmo utilizado pudieron realizarse distintas pruebas de diagnóstico molecular, a saber:
- 1)
Búsqueda de mutaciones puntuales en el ADNmt: se amplificaron fragmentos flanqueantes a regiones del genoma mitocondrial donde se localizan mutaciones causantes de síndromes clásicos (p. ej., 3.243, 8.344, 8.993, etc.) por PCR. Luego los amplificados purificados fueron secuenciados por método de Sanger. Detalles particulares de cada reacción individual están disponibles para ser requeridos a los autores.
- 2)
Secuenciación completa del genoma mitocondrial: se obtuvo la secuencia completa del genoma mitocondrial mediante enriquecimiento por Long Range PCR (LR-PCR) y NGS en sistemas GS-FLX 454 e Ilumina Miseq como se describe en Kauffman et al.7.
- 3)
Detección de alteraciones estructurales (deleciones/duplicaciones) del ADNmt: se amplificaron por LR-PCR 3 fragmentos, que solapados abarcan en su totalidad el ADNmt. Los productos obtenidos fueron analizados por electroforesis en gel de agarosa al 0,9% para evaluar visualmente la presencia de deleciones o duplicaciones en el ADNmt.
- 4)
Búsqueda de mutaciones puntuales en el ADNn: se amplificaron fragmentos flanqueantes a regiones del genoma nuclear donde se localizan mutaciones causantes en genes nucleares de síndromes clásicos (p. ej., C10orf2, POLG, etc.) por PCR. Luego los amplificados purificados fueron secuenciados por método de Sanger. Detalles particulares de cada reacción individual están disponibles para ser requeridos a los autores.
- 5)
Secuenciación completa del exoma humano: a partir de 1 μg de ADN se construyó una biblioteca de secuenciación mediante fragmentación de la muestra por nebulización en tamaños de 350-400 pb. Posteriormente, se enriqueció la biblioteca con aquellos fragmentos representativos del exoma humano completo mediante la utilización de un sistema de hibridación en solución (Nimblegen V3). Se amplificaron por PCR todos los fragmentos seleccionados y finalmente, se secuenció el exoma utilizando un equipo Illumina Hiseq 2000. Además de identificarse variantes en el genoma nuclear, se extrajo secuencia del genoma mitocondrial e se identificaron variantes en el mismo siguiendo procedimientos descriptos en Picardi et al.8.
Se utilizaron las siguientes herramientas bioinformáticas para la caracterización de las secuencias obtenidas e inferencia de patogenicidad: ENSEMBL9, MUTATION @A GLANCE10, SIFT11, POLYPHEN212, MUTATION TASTER13,14, gsMapper (Roche®), Tablet15, MITOMAP16 y PHYLOTREE17. Para el procesamiento y análisis de los exomas se utilizaron procedimientos desarrollados por nuestro grupo y descriptos en Cordoba et al.18.
Análisis estadísticoLas distintas herramientas bioinformáticas utilizadas involucran el uso de algoritmos particulares que incluyen pruebas estadísticas que habitualmente son no paramétricas y basadas en permutaciones. Una descripción detallada de cada algoritmo puede encontrarse en cada una de las referencias de las herramientas mencionadas arriba.
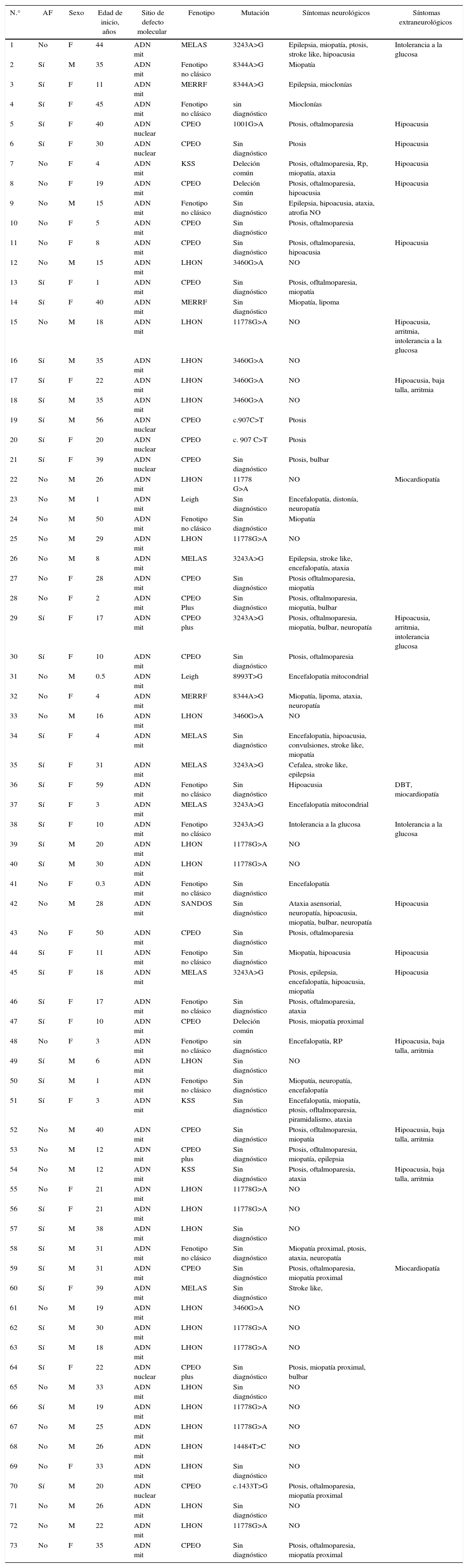
ResultadosCohorteEntre mayo del 2008 y junio del 2016 se incluyó en este estudio a un total de 73 pacientes con sospecha de desorden mitocondrial, de los cuales 39 fueron esporádicos, mientras que 34 presentaban antecedentes en la familia de trastornos similares (mitocondrial: 27 pacientes, mendeliana AD: 7 pacientes). Hubo una similar representación de sexos, donde 38 fueron mujeres. La edad promedio de inicio de los síntomas fue de 22 años, con un mínimo de 3 meses y un máximo de 59 años (tabla 1). Sesenta y un pacientes presentaron fenotipo compatible con un síndrome mitocondrial clásico, el más frecuente fue LHON (24 pacientes), seguido de CPEO (17 pacientes), MELAS (7 pacientes), CPEO Plus (4 pacientes), KSS (3 pacientes), MERRF (3 pacientes), Leigh (2 paciente) y SANDOS (un paciente) (tabla 1 y tabla 2).
Caracterización clínica de nuestra cohorte
| N.° | AF | Sexo | Edad de inicio, años | Sitio de defecto molecular | Fenotipo | Mutación | Síntomas neurológicos | Síntomas extraneurológicos |
|---|---|---|---|---|---|---|---|---|
| 1 | No | F | 44 | ADN mit | MELAS | 3243A>G | Epilepsia, miopatía, ptosis, stroke like, hipoacusia | Intolerancia a la glucosa |
| 2 | Sí | M | 35 | ADN mit | Fenotipo no clásico | 8344A>G | Miopatía | |
| 3 | Sí | F | 11 | ADN mit | MERRF | 8344A>G | Epilepsia, mioclonías | |
| 4 | Sí | F | 45 | ADN mit | Fenotipo no clásico | sin diagnóstico | Mioclonías | |
| 5 | Sí | F | 40 | ADN nuclear | CPEO | 1001G>A | Ptosis, oftalmoparesia | Hipoacusia |
| 6 | Sí | F | 30 | ADN nuclear | CPEO | Sin diagnóstico | Ptosis | Hipoacusia |
| 7 | No | F | 4 | ADN mit | KSS | Deleción común | Ptosis, oftalmoparesia, Rp, miopatía, ataxia | Hipoacusia |
| 8 | No | F | 19 | ADN mit | CPEO | Deleción común | Ptosis, oftalmoparesia, hipoacusia | Hipoacusia |
| 9 | No | M | 15 | ADN mit | Fenotipo no clásico | Sin diagnóstico | Epilepsia, hipoacusia, ataxia, atrofia NO | |
| 10 | No | F | 5 | ADN mit | CPEO | Sin diagnóstico | Ptosis, oftalmoparesia | |
| 11 | No | F | 8 | ADN mit | CPEO | Sin diagnóstico | Ptosis, oftalmoparesia, hipoacusia | Hipoacusia |
| 12 | No | M | 15 | ADN mit | LHON | 3460G>A | NO | |
| 13 | Sí | F | 1 | ADN mit | CPEO | Sin diagnóstico | Ptosis, ofltalmoparesia, miopatía | |
| 14 | Sí | F | 40 | ADN mit | MERRF | Sin diagnóstico | Miopatía, lipoma | |
| 15 | No | M | 18 | ADN mit | LHON | 11778G>A | NO | Hipoacusia, arritmia, intolerancia a la glucosa |
| 16 | Sí | M | 35 | ADN mit | LHON | 3460G>A | NO | |
| 17 | Sí | F | 22 | ADN mit | LHON | 3460G>A | NO | Hipoacusia, baja talla, arritmia |
| 18 | Sí | M | 35 | ADN mit | LHON | 3460G>A | NO | |
| 19 | Sí | M | 56 | ADN nuclear | CPEO | c.907C>T | Ptosis | |
| 20 | Sí | F | 20 | ADN nuclear | CPEO | c. 907 C>T | Ptosis | |
| 21 | Sí | F | 39 | ADN nuclear | CPEO | Sin diagnóstico | Ptosis, bulbar | |
| 22 | No | M | 26 | ADN mit | LHON | 11778 G>A | NO | Miocardiopatía |
| 23 | No | M | 1 | ADN mit | Leigh | Sin diagnóstico | Encefalopatía, distonía, neuropatía | |
| 24 | No | M | 50 | ADN mit | Fenotipo no clásico | Sin diagnóstico | Miopatía | |
| 25 | No | M | 29 | ADN mit | LHON | 11778G>A | NO | |
| 26 | No | M | 8 | ADN mit | MELAS | 3243A>G | Epilepsia, stroke like, encefalopatía, ataxia | |
| 27 | No | F | 28 | ADN mit | CPEO | Sin diagnóstico | Ptosis ofltalmoparesia, miopatía | |
| 28 | No | F | 2 | ADN mit | CPEO Plus | Sin diagnóstico | Ptosis, ofltalmoparesia, miopatía, bulbar | |
| 29 | Sí | F | 17 | ADN mit | CPEO plus | 3243A>G | Ptosis, ofltalmoparesia, miopatía, bulbar, neuropatía | Hipoacusia, arritmia, intolerancia glucosa |
| 30 | Sí | F | 10 | ADN mit | CPEO | Sin diagnóstico | Ptosis, oftalmoparesia | |
| 31 | No | M | 0.5 | ADN mit | Leigh | 8993T>G | Encefalopatía mitocondrial | |
| 32 | No | F | 4 | ADN mit | MERRF | 8344A>G | Miopatía, lipoma, ataxia, neuropatía | |
| 33 | No | M | 16 | ADN mit | LHON | 3460G>A | NO | |
| 34 | Sí | F | 4 | ADN mit | MELAS | Sin diagnóstico | Encefalopatía, hipoacusia, convulsiones, stroke like, miopatía | |
| 35 | Sí | F | 31 | ADN mit | MELAS | 3243A>G | Cefalea, stroke like, epilepsia | |
| 36 | Sí | F | 59 | ADN mit | Fenotipo no clásico | Sin diagnóstico | Hipoacusia | DBT, miocardiopatía |
| 37 | Sí | F | 3 | ADN mit | MELAS | 3243A>G | Encefalopatía mitocondrial | |
| 38 | Sí | F | 10 | ADN mit | Fenotipo no clásico | 3243A>G | Intolerancia a la glucosa | Intolerancia a la glucosa |
| 39 | Sí | M | 20 | ADN mit | LHON | 11778G>A | NO | |
| 40 | Sí | M | 30 | ADN mit | LHON | 11778G>A | NO | |
| 41 | No | F | 0.3 | ADN mit | Fenotipo no clásico | Sin diagnóstico | Encefalopatía | |
| 42 | No | M | 28 | ADN mit | SANDOS | Sin diagnóstico | Ataxia asensorial, neuropatía, hipoacusia, miopatía, bulbar, neuropatía | Hipoacusia |
| 43 | No | F | 50 | ADN mit | CPEO | Sin diagnóstico | Ptosis, oftalmoparesia | |
| 44 | Sí | F | 11 | ADN mit | Fenotipo no clásico | Sin diagnóstico | Miopatía, hipoacusia | Hipoacusia |
| 45 | Sí | F | 18 | ADN mit | MELAS | 3243A>G | Ptosis, epilepsia, encefalopatía, hipoacusia, miopatía | Hipoacusia |
| 46 | Sí | F | 17 | ADN mit | Fenotipo no clásico | Sin diagnóstico | Ptosis, oftalmoparesia, ataxia | |
| 47 | Sí | F | 10 | ADN mit | CPEO | Deleción común | Ptosis, miopatía proximal | |
| 48 | No | F | 3 | ADN mit | Fenotipo no clásico | sin diagnóstico | Encefalopatía, RP | Hipoacusia, baja talla, arritmia |
| 49 | Sí | M | 6 | ADN mit | LHON | Sin diagnóstico | NO | |
| 50 | Sí | M | 1 | ADN mit | Fenotipo no clásico | Sin diagnóstico | Miopatía, neuropatía, encefalopatía | |
| 51 | Sí | F | 3 | ADN mit | KSS | Sin diagnóstico | Encefalopatía, miopatía, ptosis, ofltalmoparesia, piramidalismo, ataxia | |
| 52 | No | M | 40 | ADN mit | CPEO | Sin diagnóstico | Ptosis, ofltalmoparesia, miopatía | Hipoacusia, baja talla, arritmia |
| 53 | No | M | 12 | ADN mit | CPEO plus | Sin diagnóstico | Ptosis, ofltalmoparesia, miopatía, epilepsia | |
| 54 | No | M | 12 | ADN mit | KSS | Sin diagnóstico | Ptosis, oftalmoparesia, ataxia | Hipoacusia, baja talla, arritmia |
| 55 | No | F | 21 | ADN mit | LHON | 11778G>A | NO | |
| 56 | Sí | F | 21 | ADN mit | LHON | 11778G>A | NO | |
| 57 | Sí | M | 38 | ADN mit | LHON | Sin diagnóstico | NO | |
| 58 | Sí | M | 31 | ADN mit | Fenotipo no clásico | Sin diagnóstico | Miopatía proximal, ptosis, ataxia, neuropatía | |
| 59 | Sí | M | 31 | ADN mit | CPEO | Sin diagnóstico | Ptosis, oftalmoparesia, miopatía proximal | Miocardiopatía |
| 60 | Sí | F | 39 | ADN mit | MELAS | Sin diagnóstico | Stroke like, | |
| 61 | No | M | 19 | ADN mit | LHON | 3460G>A | NO | |
| 62 | Sí | M | 30 | ADN mit | LHON | 11778G>A | NO | |
| 63 | Sí | M | 18 | ADN mit | LHON | 11778G>A | NO | |
| 64 | Sí | F | 22 | ADN nuclear | CPEO plus | Sin diagnóstico | Ptosis, miopatía proximal, bulbar | |
| 65 | No | M | 33 | ADN mit | LHON | Sin diagnóstico | NO | |
| 66 | Sí | M | 19 | ADN mit | LHON | 11778G>A | NO | |
| 67 | No | M | 25 | ADN mit | LHON | 11778G>A | NO | |
| 68 | No | M | 26 | ADN mit | LHON | 14484T>C | NO | |
| 69 | No | F | 33 | ADN mit | LHON | Sin diagnóstico | NO | |
| 70 | Sí | M | 20 | ADN nuclear | CPEO | c.1433T>G | Ptosis, oftalmoparesia, miopatía proximal | |
| 71 | No | M | 26 | ADN mit | LHON | Sin diagnóstico | NO | |
| 72 | No | M | 22 | ADN mit | LHON | 11778G>A | NO | |
| 73 | No | F | 35 | ADN mit | CPEO | Sin diagnóstico | Ptosis, oftalmoparesia, miopatía proximal |
AD: autosómico dominante; CPEO: Chronic progressive external ophthalmoplegia; KSS: Kearns-Sayre syndrome; LHON: Leber hereditary optic neuropathy; MELAS: Mitochondrial encephalomyopathy, lactic acidosis, and stroke-like episodes; MERRF: Myoclonic Epilepsy Associated with Ragged Red Fibers; NO: neuropatía óptica.
La manifestación neurológica más frecuente fue la debilidad muscular en miembros (26 pacientes), seguida de neuropatía óptica bilateral (25 pacientes), ptosis-oftalmoparesia (21 pacientes), encefalopatía (11 pacientes), ptosis aislada (9 pacientes), epilepsia (9 pacientes), ataxia (8 pacientes), episodios stroke like (5 pacientes), neuropatía (5 pacientes), disfagia con otros síntomas bulbares (4 pacientes), mioclonías (3 pacientes), retinopatía pigmentaria (2 pacientes), y con mucho menos frecuencia cefalea y distonía en un paciente, respectivamente. Entre las manifestaciones extraneurológicas se incluye la sordera neurosensorial bilateral en 16 pacientes, las arritmias (6 pacientes), la intolerancia a la glucosa (5 pacientes), las cardiomiopatías (4 pacientes), los lipomas (2 pacientes), baja talla (un paciente) y hepatopatía en un paciente (tabla 2).
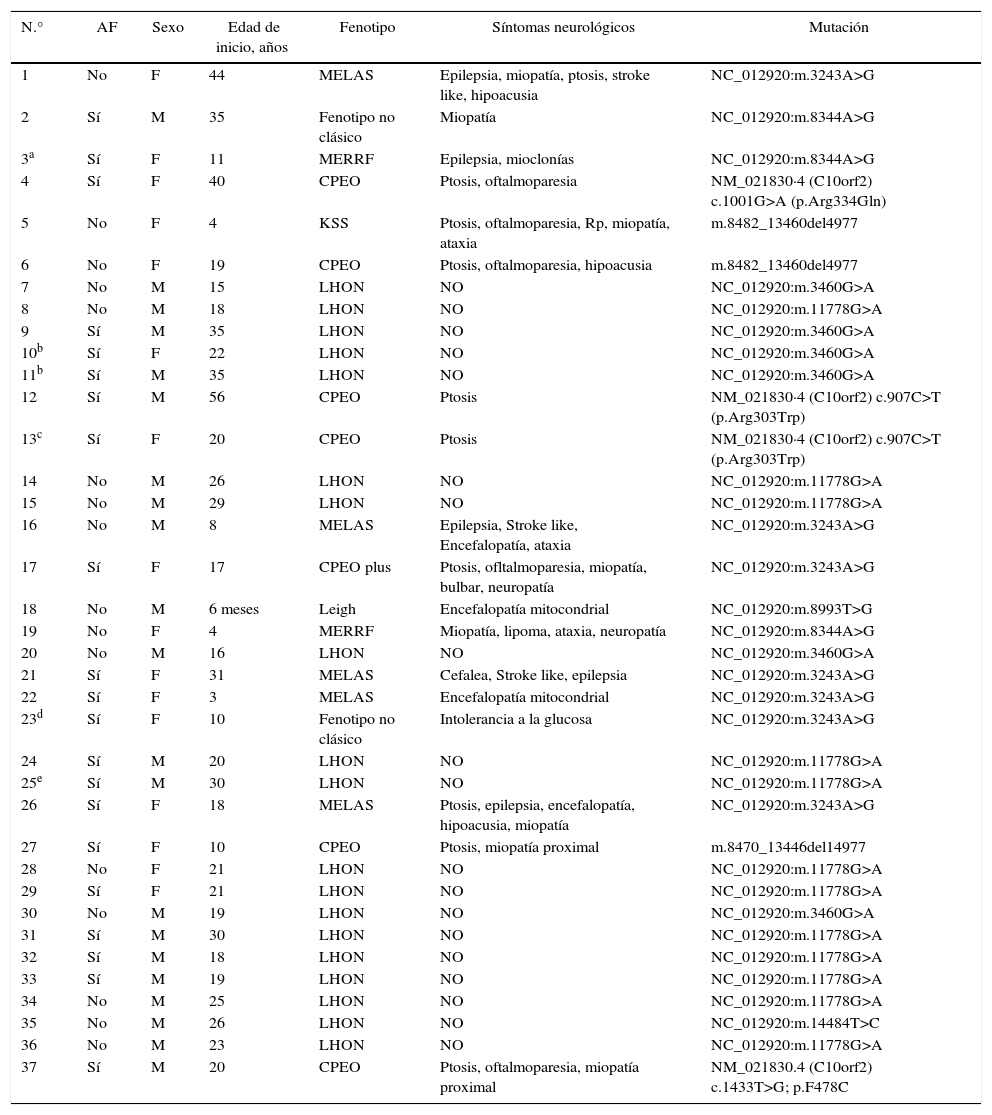
Rendimiento diagnóstico molecularSiguiendo el algoritmo propuesto, obtuvimos un rendimiento diagnóstico molecular de un 51%, pudiendo identificar alteraciones en 37 pacientes. Mutaciones puntuales en el ADNmt fueron individualizadas en 30 pacientes. Alteraciones estructurales en el genoma mitocondrial en 3 y mutaciones en genes nucleares en 4 (tablas 3 y 4).
Detalle de los pacientes en los que fue identificado un defecto molecular y su presentación fenotípica. Referencias:
| N.° | AF | Sexo | Edad de inicio, años | Fenotipo | Síntomas neurológicos | Mutación |
|---|---|---|---|---|---|---|
| 1 | No | F | 44 | MELAS | Epilepsia, miopatía, ptosis, stroke like, hipoacusia | NC_012920:m.3243A>G |
| 2 | Sí | M | 35 | Fenotipo no clásico | Miopatía | NC_012920:m.8344A>G |
| 3a | Sí | F | 11 | MERRF | Epilepsia, mioclonías | NC_012920:m.8344A>G |
| 4 | Sí | F | 40 | CPEO | Ptosis, oftalmoparesia | NM_021830·4 (C10orf2) c.1001G>A (p.Arg334Gln) |
| 5 | No | F | 4 | KSS | Ptosis, oftalmoparesia, Rp, miopatía, ataxia | m.8482_13460del4977 |
| 6 | No | F | 19 | CPEO | Ptosis, oftalmoparesia, hipoacusia | m.8482_13460del4977 |
| 7 | No | M | 15 | LHON | NO | NC_012920:m.3460G>A |
| 8 | No | M | 18 | LHON | NO | NC_012920:m.11778G>A |
| 9 | Sí | M | 35 | LHON | NO | NC_012920:m.3460G>A |
| 10b | Sí | F | 22 | LHON | NO | NC_012920:m.3460G>A |
| 11b | Sí | M | 35 | LHON | NO | NC_012920:m.3460G>A |
| 12 | Sí | M | 56 | CPEO | Ptosis | NM_021830·4 (C10orf2) c.907C>T (p.Arg303Trp) |
| 13c | Sí | F | 20 | CPEO | Ptosis | NM_021830·4 (C10orf2) c.907C>T (p.Arg303Trp) |
| 14 | No | M | 26 | LHON | NO | NC_012920:m.11778G>A |
| 15 | No | M | 29 | LHON | NO | NC_012920:m.11778G>A |
| 16 | No | M | 8 | MELAS | Epilepsia, Stroke like, Encefalopatía, ataxia | NC_012920:m.3243A>G |
| 17 | Sí | F | 17 | CPEO plus | Ptosis, ofltalmoparesia, miopatía, bulbar, neuropatía | NC_012920:m.3243A>G |
| 18 | No | M | 6 meses | Leigh | Encefalopatía mitocondrial | NC_012920:m.8993T>G |
| 19 | No | F | 4 | MERRF | Miopatía, lipoma, ataxia, neuropatía | NC_012920:m.8344A>G |
| 20 | No | M | 16 | LHON | NO | NC_012920:m.3460G>A |
| 21 | Sí | F | 31 | MELAS | Cefalea, Stroke like, epilepsia | NC_012920:m.3243A>G |
| 22 | Sí | F | 3 | MELAS | Encefalopatía mitocondrial | NC_012920:m.3243A>G |
| 23d | Sí | F | 10 | Fenotipo no clásico | Intolerancia a la glucosa | NC_012920:m.3243A>G |
| 24 | Sí | M | 20 | LHON | NO | NC_012920:m.11778G>A |
| 25e | Sí | M | 30 | LHON | NO | NC_012920:m.11778G>A |
| 26 | Sí | F | 18 | MELAS | Ptosis, epilepsia, encefalopatía, hipoacusia, miopatía | NC_012920:m.3243A>G |
| 27 | Sí | F | 10 | CPEO | Ptosis, miopatía proximal | m.8470_13446del14977 |
| 28 | No | F | 21 | LHON | NO | NC_012920:m.11778G>A |
| 29 | Sí | F | 21 | LHON | NO | NC_012920:m.11778G>A |
| 30 | No | M | 19 | LHON | NO | NC_012920:m.3460G>A |
| 31 | Sí | M | 30 | LHON | NO | NC_012920:m.11778G>A |
| 32 | Sí | M | 18 | LHON | NO | NC_012920:m.11778G>A |
| 33 | Sí | M | 19 | LHON | NO | NC_012920:m.11778G>A |
| 34 | No | M | 25 | LHON | NO | NC_012920:m.11778G>A |
| 35 | No | M | 26 | LHON | NO | NC_012920:m.14484T>C |
| 36 | No | M | 23 | LHON | NO | NC_012920:m.11778G>A |
| 37 | Sí | M | 20 | CPEO | Ptosis, oftalmoparesia, miopatía proximal | NM_021830.4 (C10orf2) c.1433T>G; p.F478C |
Se describen 2 casos clínicos que creemos relevantes porque compartiendo una misma mutación presentan fenotipos bien diferentes, siendo ilustrativos del pleomorfismo clínico de las enfermedades mitocondriales, su dificultad diagnóstica y la necesidad de herramientas moleculares para su diagnóstico.
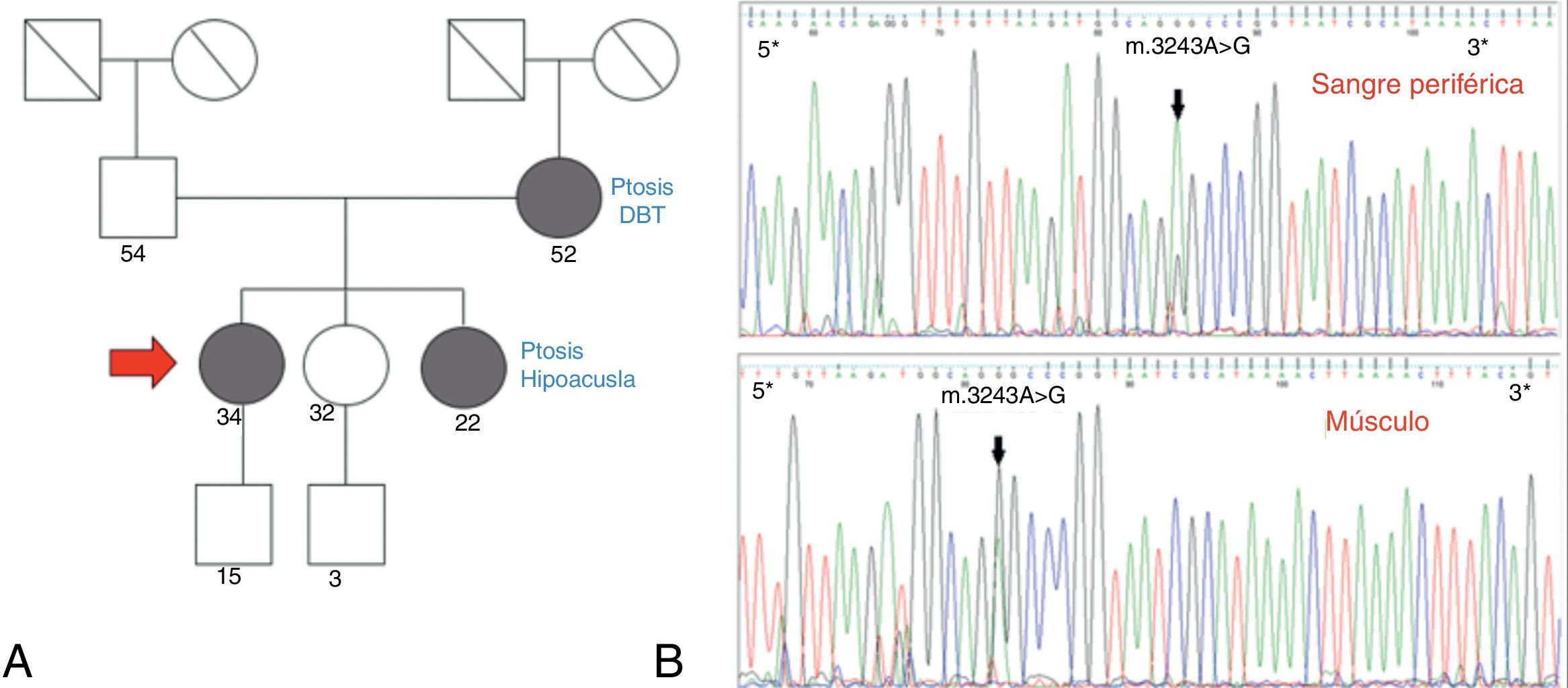
Caso 1. Paciente de 34 años, sin antecedentes personales de relevancia, que comenzó a los 17 años con ptosis palpebral bilateral y oftalmoparesia de curso lentamente progresivo. A los 30 años agregó debilidad proximal de 4 miembros y trastornos fonodeglutorios. Su historia familiar era relevante por la presencia de ptosis bilateral en su madre y hermana.
En el examen físico presentaba ptosis palpebral bilateral 2/3, oftalmoparesia moderada, voz nasal. Hipoacusia bilateral. Debilidad proximal de 4 miembros leve (Kendall 3-4/5). Sensibilidad conservada. Normorreflexia generalizada. Los exámenes complementarios confirmaron además la presencia de: hipoacusia neurosensorial, intolerancia a la glucosa, trastorno de la conducción supraventricular cardiaca, aumento de creatincinasa y ácido láctico en sangre. El fenotipo fue interpretado como compatible con CPEO Plus.
Se realizó biopsia de músculo, la cual evidenció alto porcentaje de FRR, Cox deficientes. Se realizó secuenciación por Sanger de ADNmt, hallándose la mutación para NC_012920:m.3243A>G tanto en sangre periférica como en músculo, por lo que se infiere un alto nivel de heteroplasmia (fig. 3).
 Familigrama. B) Secuenciación por Sanger en sangre periférica y en músculo evidenciando mutación NC_012920:m.3243.")
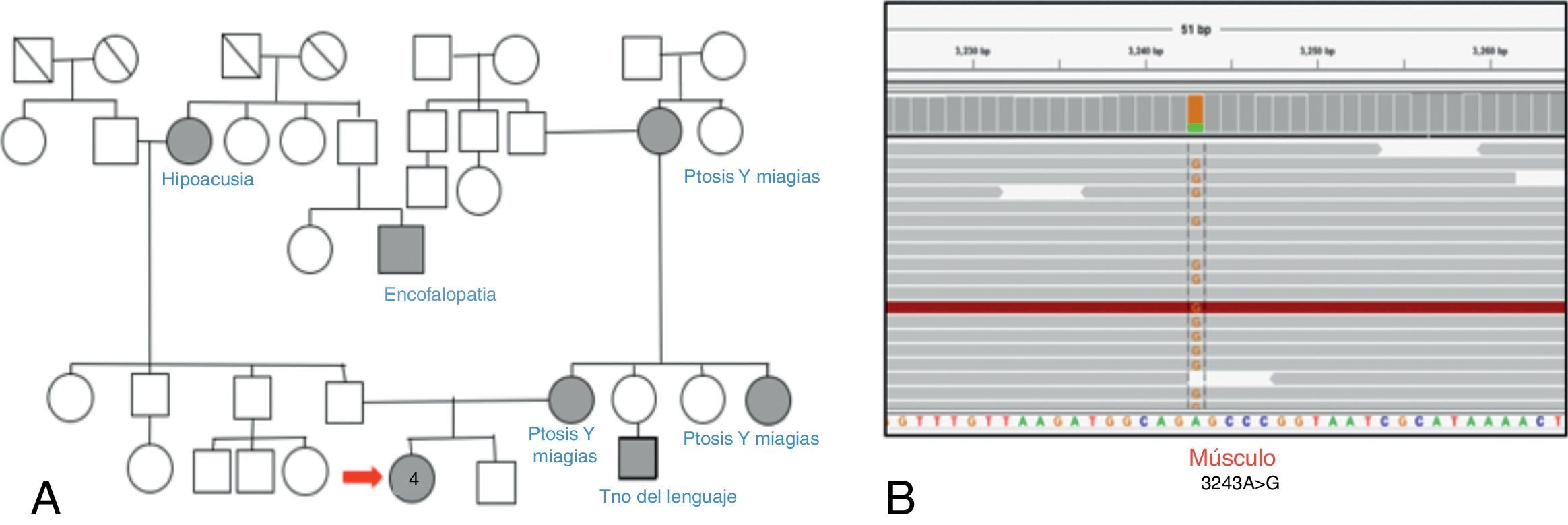
Caso 2. Paciente de 26 años, sin antecedentes personales o familiares de relevancia; comenzó a los 8 años con trastornos del aprendizaje, 12 años después agregó ataxia de tronco con lateropulsión a derecha, trastornos conductuales e intolerancia al ejercicio. A los 25 años presentó disartria y hemianopsia homónima derecha asociada a crisis comicial focal motora en el miembro superior derecho. Se le realizó RM de cerebro que evidenció lesión córtico-subcortical parietotemporooccipital izquierda con restricción en difusión, la cual remitió espontáneamente y en forma completa al mes, por lo que se interpretó como episodio «stroke like».
En el examen físico presentaba MiniMental State Examination de 23/30, hipoacusia bilateral, dismetría apendicular de 4 miembros de predominio derecho. Los exámenes complementarios confirmaron además la presencia de: hipoacusia neurosensorial y aumento de ácido láctico en sangre y líquido cefalorraquídeo. El fenotipo fue interpretado como compatible con MELAS.
Se realizó biopsia de músculo, la cual evidenció un alto porcentaje de FRR, Cox deficientes. Se realizó secuenciación por Sanger de ADNmt que puso de manifiesto también en este caso la mutación NC_012920:m.3243A>G en músculo. Se suspendió el ácido valproico y se rotó medicación antiepiléptica a levetiracetam con buen control de la crisis (fig. 4).
Discusión Familigrama. B) Secuenciación por Sanger en músculo evidenciando mutación NC_012920:m.3243.")
En el presente trabajo pudimos caracterizar una población adulta de pacientes argentinos con enfermedad mitocondrial implementando un algoritmo propio que pretende ordenar la aproximación diagnóstica, comenzando con un score de probabilidad de etiología mitocondrial del trastorno, siguiendo por una caracterización sindrómica y finalizando con el diagnóstico molecular.
Hasta donde sabemos, esta es la serie más grande reportada en Argentina que incluye datos clínicos y moleculares, estos últimos obtenidos a través del desarrollo propio de diferentes metodologías que incorporan el estudio de genoma mitocondrial y de exoma completo mediante NGS.
A pesar de que las enfermedades mitocondriales se caracterizan por un marcado pleomorfismo clínico y una escasa correlación fenotipo-genotipo1, pudimos identificar el defecto causal en 37 pacientes. Nuestros resultados confirman la utilidad del algoritmo diagnóstico propuesto expresada en un alto rendimiento diagnóstico (51%), que deriva en mejor costo-efectividad. A modo de ejemplo, a través de la secuenciación de exoma completo por NGS y extracción del genoma mitocondrial utilizando un pipeline especializado fue posible la identificación de una mutación en el ADNmt no sospechada previamente por el fenotipo en una niña con presentación en forma de síndrome no clásico. La utilización de NGS y LR-PCR nos permitió aumentar la tasa diagnóstica comparando con nuestros propios resultados obtenidos previamente utilizando solamente secuenciación dirigida de fragmento único mediante método de Sanger19.
Si bien existen otras series reportadas en la literatura, un gran número de ellas analizaron muestras infantiles, por lo que las manifestaciones clínicas más frecuentes incluyeron encefalopatía y trastornos del aprendizaje, seguidas de debilidad muscular20. Nuestra serie difiere en la inclusión mayoritaria de adultos con un rango de edad más grande, siendo más frecuente la presentación sindromática clásica que la no clásica, con un predominio de LHON, CPEO y MELAS. Distintos artículos publicados a lo largo de toda una década revelaron una dicotomía molecular entre adultos y niños, en la que los niños frecuentemente portan trastornos autosómicos recesivos debidos a defectos del ADNn, mientras que los defectos del ADNmt se presentan más frecuentemente en la edad adulta21,22. En los últimos años esta separación se ha puesto en revisión, a la luz de que las mutaciones nucleares con herencia mendeliana pueden ocurrir frecuentemente también en adultos. De ahí la importancia de aportar conocimiento científico en este grupo etario incluyendo el estudio de genes mitocondriales del genoma nuclear, tal como hicimos en nuestro trabajo23.
Los avances en el diagnóstico molecular de las enfermedades mitocondriales han permitido dilucidar nuevos mecanismos en el desarrollo de los fenotipos clínicos que pueden estar vinculados a la función primaria de la proteína mutada, a la acumulación secundaria de deleciones y mutaciones puntuales o a la depleción del ADNmt. Esto abre las puertas a nuevas terapéuticas orientadas a prevenir y reparar los daños del ADNmt, que podrían ser aplicadas en distintas etapas de la enfermedad23.
Concluimos que una aproximación ordenada y sistematizada, como la que proponemos, permite confirmar el diagnóstico molecular en un porcentaje alto de pacientes. Esto resulta de gran valor para una mejor comprensión de la patogénesis de las enfermedades neurogenéticas y para una asistencia médica más eficiente e integral de los pacientes y sus familias.
Responsabilidades éticasProtección de personas y animalesLos autores declaran que para esta investigación no se han realizado experimentos en seres humanos ni en animales.
Confidencialidad de los datosLos autores declaran que han seguido los protocolos de su centro de trabajo sobre la publicación de datos de pacientes.
Derecho a la privacidad y consentimiento informadoLos autores declaran que en este artículo no aparecen datos de pacientes.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.
