




La encefalitis de tronco de Bickerstaff (EB) es una enfermedad autoinmune rara que afecta a estructuras de sistema nervioso central. Se caracteriza por la presencia de ataxia, alteración del sensorio y oftalmoplejía. Se han descrito casos con presentación atípica.
Métodos y resultadosPresentamos 2 casos pediátricos de EB atípica.
ConclusiónLa EB es una patología rara. Su identificación y tratamiento precoz condicionan la morbimortalidad de estos pacientes.
Brainstem Bickerstaff Encephalytis is a rare autoimmune disease that affects central nervous system structures. It is characterized by the presence of ataxia, sensory disturbance and ophthalmoplegia. There have been reportedatientes with atypical presentation.
Methods and resultsWe present two cases of atypical pediatric Bickerstaff encephalytis.
ConclusionThe Bickerstaff encephalytis is a rare disease. Early identification and treatment determine morbidity and mortality of these patients.
La encefalitis es un proceso inflamatorio del sistema nervioso central (SNC), cuya característica clínica principal es la disfunción neurológica. Se debe a múltiples agentes etiológicos, fundamentalmente virus. La contigüidad de las estructuras del SNC hace que se presenten cuadros clínicos mixtos: si la infección afecta al cerebro y a las meninges, se conoce como meningoencefalitis; si solo afecta al cerebro como encefalitis y si afecta a la médula espinal mielitis o encefalomielitis. Las tasas de encefalitis varía de 3 casos por cada 100.000 personas a 33 por cada 100.000 personas al año, siendo mayores las cifras en el caso de los niños. La probabilidad del desarrollo de la encefalitis y su gravedad dependerá tanto del medio de adquisición (contacto con humanos o del entorno) como de la edad, la inmunocompetencia del huésped y la disponibilidad de vacunas1..
La encefalitis de tallo cerebral de Bickerstaff es un síndrome definido por la presencia de ataxia con oftalmoplejía asociada a alteración del sensorio o reflejos exaltados. Desde su primera descripción en 1950, esta entidad se ha convertido en un reto diagnóstico, así como en un tema de debate entre los neurólogos clínicos. Se considera una patología con base autoinmune. Parece existir una posible asociación entre esta entidad y el síndrome de Miller Fisher (SMF) descrito en 1956 y, por ende, con el síndrome de Guillain-Barré (SGB), aunque todavía no está clara la interrelación1,2.
Presentamos 2 casos de niños varones con edades comprendidas entre los 12 y los 24 meses de edad, diagnosticados y tratados en nuestro hospital con cuadros compatibles con romboencefalitis.
Casos clínicosCaso 1Varón de 2 años que presenta vómitos de predominio matutino, cefalea intermitente y fiebre (máx. 38,7°C) de 72 h de evolución. Acude al Servicio de Urgencias por inestabilidad de la marcha de 12 h de evolución. Hermana de 5 años, sana. El padre presenta síndrome de piernas inquietas. Resto de antecedentes personales sin interés.
En la exploración física destacan los reflejos osteotendinosos (ROT) débiles junto a marcha atáxica. Reflejo cutáneo plantar flexor bilateral en ese momento.
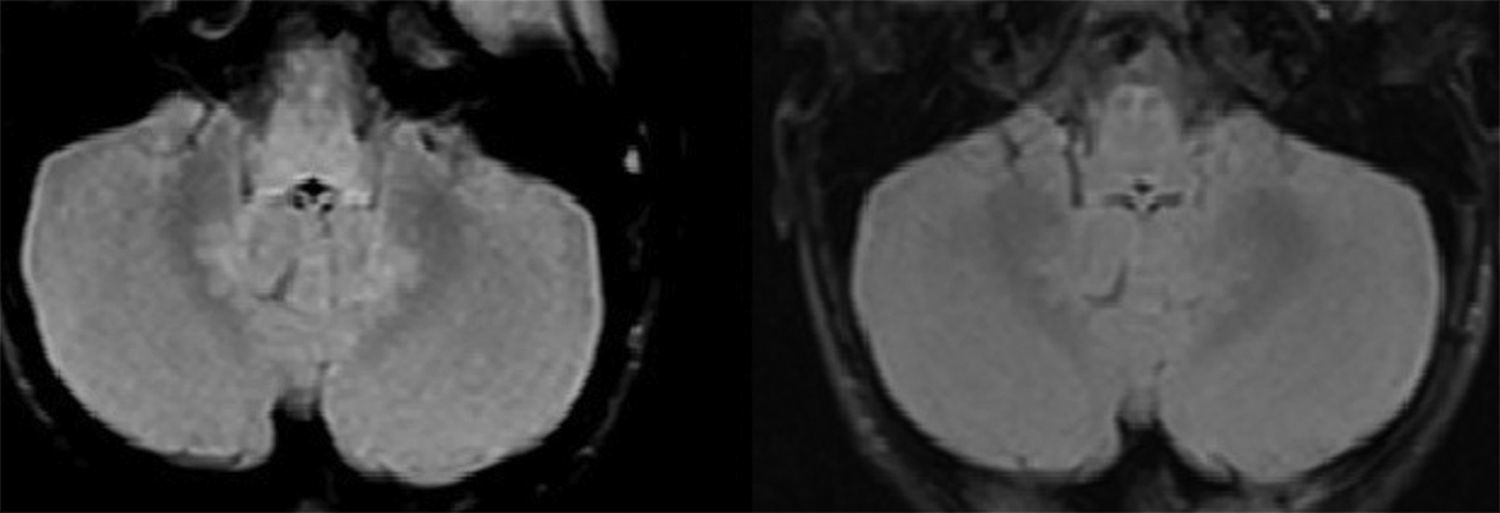
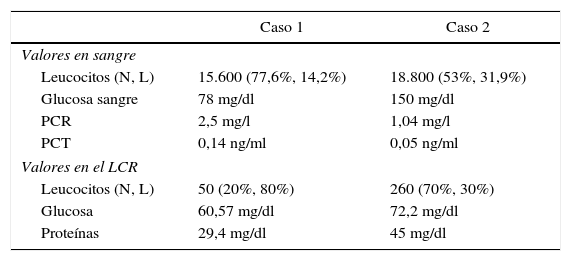
En las pruebas complementarias destaca la presencia de leucocitosis con predominio de neutrófilos en el hemograma, así como la pleocitosis en el líquido cefalorraquídeo (LCR) con predominio de mononucleares. Los reactantes de fase aguda (proteína C reactiva y procalcitonina) fueron normales (tabla 1). Se solicitaron como estudios neurofisiológicos electroencefalograma (EEG), electroneurograma (ENG), electromiograma (EMG), potenciales de tronco (PEATC) y potenciales evocados visuales (PEV). Se observó en los PEATC un retraso en la conducción central del sonido y en el EMG signos de denervación aguda del musculo tibial anterior derecho (L4-L5). La tomografía computarizada cerebral fue normal. La resonancia magnética (RM) craneal mostró alteración de la intensidad de señal de la región posterior de la protuberancia, pedúnculos cerebelosos y sustancia blanca profunda cerebelosa, bilateral y simétrica, hiperintensa en secuencias de TR largo, hipointensa en T1; no brilla ni restringe en difusión. Asimismo, se observó una sutil hiperintensidad de señal difusa desde la unión bulbomedular hasta la región central de la médula dorsal (fig. 1). Se extrajeron serologías para Lysteria, Mycoplasma, Herpes viridae, Borrelia, Salmonella typhi, enterovirus, Echovirus y parvovirus negativos. El estudio de anticuerpos (Ac) antigangliósido (GM, GD, GT, GQ) y Ac antineuronales (anti-Hu, anti-yo, anti-VGKC, anti-VGCC, anti-NMDA) también fue negativo. Durante su ingreso comienza con dificultad para la sedestación, temblor en miembros superiores, Babinski izquierdo, derecho indiferente y habla escándida. Asimismo tiene cierta tendencia al sueño, por lo que se decide iniciar tratamiento con aciclovir, ampicilina, claritromicina, inmunoglobulinas por vía intravenosa (Ig IV) y metilprednisolona IV, con mejoría clínica progresiva. Presenta una recuperación completa en 7 días. Desde el alta, el paciente está asintomático.
Valores analíticos en sangre periférica y LCR
| Caso 1 | Caso 2 | |
|---|---|---|
| Valores en sangre | ||
| Leucocitos (N, L) | 15.600 (77,6%, 14,2%) | 18.800 (53%, 31,9%) |
| Glucosa sangre | 78 mg/dl | 150 mg/dl |
| PCR | 2,5 mg/l | 1,04 mg/l |
| PCT | 0,14 ng/ml | 0,05 ng/ml |
| Valores en el LCR | ||
| Leucocitos (N, L) | 50 (20%, 80%) | 260 (70%, 30%) |
| Glucosa | 60,57 mg/dl | 72,2 mg/dl |
| Proteínas | 29,4 mg/dl | 45 mg/dl |
L: linfocitos; N: neutrófilos; PCR: proteína C reactiva; PCT procalcitonina.
.")
Varón de 12 meses de edad que ingresa por abombamiento de la fontanela y fiebre (máx. 38,1°C) de 24h de evolución. En las últimas horas asocia decaimiento, temblor y rechazo de la marcha. Una prima por parte materna tiene epilepsia. Resto de antecedentes familiares, sin interés. Se administró la vacuna triple vírica, neumococo 13-v y rotavirus una semana antes del inicio del cuadro. En la exploración física se observan signos de meningismo y se confirma la presencia de fontanela abombada, siendo el resto de la exploración normal.
En las pruebas complementarias destaca la presencia de leucocitosis con predominio de neutrófilos en el hemograma, así como la pleocitosis en el LCR, con predominio de mononucleares. Los reactantes de fase aguda (proteína C reactiva y procalcitonina) fueron normales (tabla 1). Se extraen serologías para Lysteria, Mycoplasma, Herpes viridae, Borrelia, Salmonella typhi, enterovirus, Echovirus y parvovirus, y se inicia tratamiento con cefotaxima IV. El cultivo de LCR y las serologías fueron negativos.
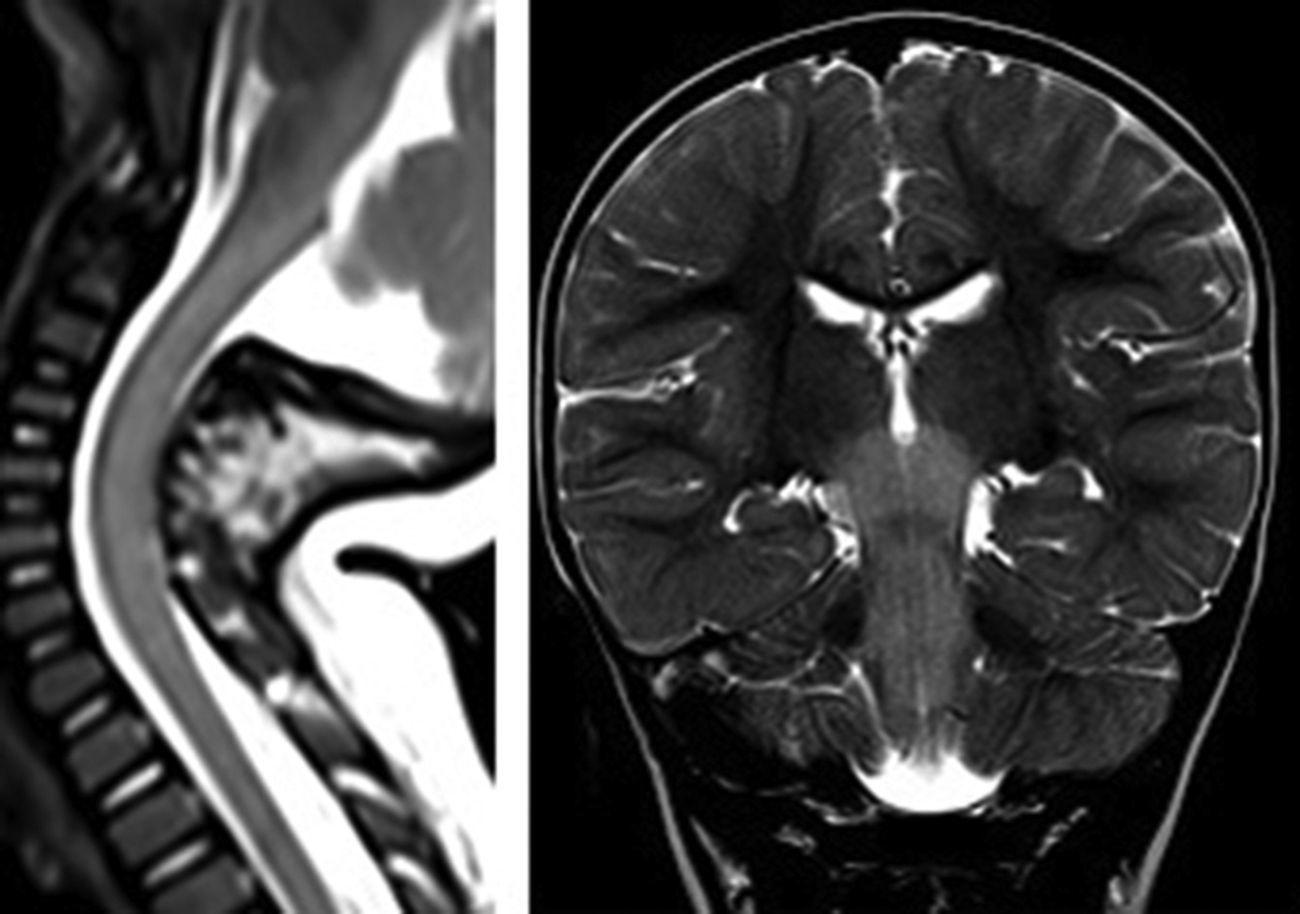
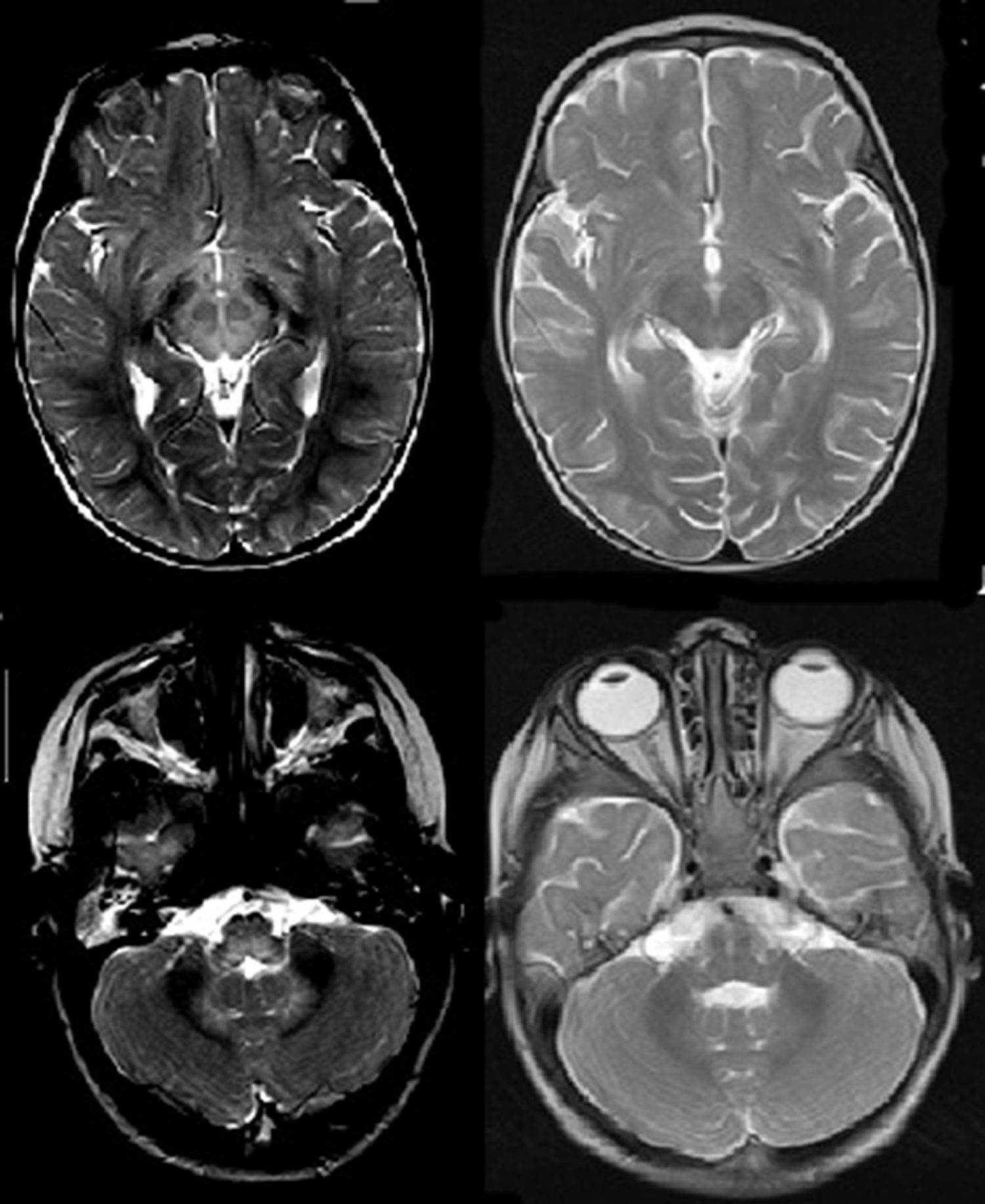
Al tercer día del ingreso, inicia un cuadro de disminución del estado de consciencia y de movimientos espontáneos, con reflejo cutáneo plantar Babinski bilateral. Ante la sospecha de encefalitis aguda, se inicia tratamiento con aciclovir. Se solicitó RM craneal que mostraba extensa afectación del romboencéfalo y médula cervical, que implicaba a toda la médula cervical, región posterior del bulbo y protuberancia, pedúnculos cerebelosos, mesencéfalo y núcleos subtalámicos. Condiciona tumefacción del tronco y aumento de grosor del cordón medular afectado. Es bilateral y simétrica (figs. 2 y 3). Se amplió estudio con EEG, ENG, EMG, PEATC y PEV, siendo los resultados normales. El estudio de Ac antigangliósido (GM, GD, GT, GQ) y Ac antineuronales (anti-Hu, anti-yo, anti-VGKC, anti-VGCC, anti-NMDA) también fue negativo. Se asoció al tratamiento previo claritromicina, ampicilina, inmunoglobulinas y corticoides IV, con mejoría clínica progresiva. Durante la recuperación del estado de consciencia se objetiva en la exploración hipotonía global con ataxia troncular. Temblor en miembros superiores. ROT abolidos en miembros superiores e inferiores y Babinski bilateral. Ausencia de coordinacion óculo-manual. No capacidad de habla ni de deglución, por lo que precisa alimentación por sonda nasogástrica. Presenta una recuperación progresiva, con exploración neurológica y desarrollo psicomotor normales a los 4 meses del inicio del cuadro.

Axial T2, a la izquierda corte axial a nivel del mesencéfalo, hiperintensidad de señal del tronco, con normalización en el control; a la derecha, corte axial a nivel del bulbo y cerebelo, hiperintensidad de señal en bulbo y núcleos dentados del cerebelo, con normalización en el control.

Las encefalitis son enfermedades inflamatorias del SNC secundarias a numerosas causas, siendo las más frecuentes las infecciones víricas. Además de cefalea, vómitos y afectación sistémica, muchos pacientes presentan alteración del sensorio, desde somnolencia hasta coma. La fiebre puede ser de bajo grado y en ocasiones aparecen también crisis convulsivas, ya sean generalizadas o parciales. La exploración física puede mostrar diversos signos clínicos como ataxia, hemiparesias, disartria… En casos graves puede haber signos de hipertensión intracraneal. El LCR en estos pacientes presenta linfocitosis, con aumento medio de proteínas y una glucorraquia normal o ligeramente disminuida. En ocasiones, si el estudio se realiza en estadios iniciales, puede aparecer predominio de neutrófilos. La neuroimagen tiene una importancia primordial en la evaluación de pacientes con encefalitis, ya que puede orientar a la etiología la encefalitis. Así la encefalitis por herpes simple muestra lesiones en el lóbulo temporal o en el caso de encefalitis por virus de Epstein-Barr aparecen lesiones en los ganglios basales. El diagnóstico microbiológico incluye estudios serológicos, cultivos celulares, estudios inmunohistoquímicos y métodos moleculares como la proteína C reactiva. Finalmente, se solicitarán estudios neurofisiológicos (EEG, EMG/ENG, PEV, etc.) en función de la sospecha clínica1.
Sin embargo, en ocasiones, aunque el desencadenante del proceso pueda ser infeccioso, la encefalitis presenta una etiología autoinmune. Existen diversas enfermedades en las que la lesión es producida por una disfunción del sistema inmunitario, no por el agente infeccioso en sí. El SMF, la encefalomielitis aguda diseminada (EMAD) o el SGB son algunos de dichos cuadros autoinmunes que en muchas ocasiones comienzan relacionados con una infección concomitante; es el caso de la infección por Campylobacter spp. y SGB. En 1957 Bickerstaff definió el síndrome de la «encefalitis de tronco o encefalitis de Bickerstaff (EB)». Se caracteriza por la presencia de ataxia con oftalmoplejía asociada a alteración del sensorio. Se clasifica dentro del espectro en enfermedad del SGB y el SMF, ya que comparten características similares. Todas estas entidades suelen precederse de pródromos infecciosos (normalmente un cuadro catarral). En un gran número de pacientes se ha detectado aumento de anticuerpos IgG antigangliósido, siendo el más frecuentemente detectado el anti-GQ1b pero también otros (GD1b, GM1, GM2…). También se ha encontrado disociación albúmino-citológica en el LCR, que implicaría una lesión de la barrera hematoencefálica. Los Ac anti-GQ1b, así como el anti-GM1, se han descrito en casos de SGB, SMF, EB y oftalmoparesia aguda sin ataxia. Sin embargo, a día de hoy todavía no está esclarecido si, efectivamente, la EB forma parte del espectro de enfermedad del SGB-SMF o si son entidades diferentes. En ocasiones, la EB puede asociar un SGB con afectación de nervio periférico con clínica y alteraciones neurofisiológicas propias de este, como es la debilidad de las extremidades de tipo flácido y/o la ausencia de ROT2-5.
Los criterios diagnósticos de la EB incluyen oftalmoplejía progresiva y ataxia, más o menos simétrica, de 4 semanas de duración, junto a reflejos exaltados o alteración del sensorio. Predomina en el sexo masculino y su edad de presentación es bimodal, con un pico inicial entre los 20 y 29 años y, posteriormente, entre los 40 y 49 años. La aparición en pacientes pediátricos es infrecuente. Es necesario descartar otras entidades que pueden dar lugar a una clínica similar, como son la miastenia gravis, la encefalomielitis aguda diseminada, el botulismo, el infarto cerebral de tronco del encéfalo, las neoplasias o la enfermedad de Wernicke, entre otras. El grupo de Masaaki Odata et al. hizo una revisión de 62 casos de pacientes con EB. Durante la enfermedad, los pacientes presentaron diferentes síntomas y signos neurológicos, además de los establecidos en los criterios diagnósticos, como blefaroptosis, debilidad de extremidades, nistagmo, debilidad facial, parálisis bulbar, alteración de ROT (desde ausentes hasta exaltados), presencia de signo de Babinski y/o alteración de la sensibilidad superficial y/o profunda. Los dividieron en un grupo con debilidad de extremidades y otro sin él, considerando que el primer grupo presentaba una EB asociado a un SGB. Las manifestaciones clínicas y prodrómicas de ambos grupos fueron muy similares2,6,7.
Se cree que el desencadenante es un agente infeccioso. El mimetismo molecular daría lugar a una reacción inmunitaria inadecuada lesionando tejidos del sistema nervioso. Se han descrito casos de EB en relación con infección por Campylobacter jejuni, varicela zóster, citomegalovirus, virus del sarampión, Lysteria monocytogenes… En el caso 2, el paciente había recibido la vacuna de la triple vírica (sarampión, rubéola y parotiditis) 7 días antes. En la serie de Masaaki Odata et al. se encontraron lesiones hiperintensas en T2 en la RM en menos del 50% de los pacientes localizadas en tronco del encéfalo, tálamos, cerebelo o sustancia blanca supratentorial. En nuestros casos, ambos pacientes presentaban lesiones de tronco del encéfalo y/o cerebelo (figs. 1-3). En los estudios de ENG el patrón predominante fue el de degeneración axonal y en el EMG de pacientes con debilidad de extremidades encontraron potenciales de denervación activos, hallazgo encontrado en nuestro caso 2. Asimismo encontraron anomalías en los EEG, aunque no en todos los casos. Los hallazgos del LCR fueron variables pero lo más frecuente fue el aumento de la proteinorraquia, leucocitosis y disociación albúmino-citológica. En muchos casos los pacientes presentan autoanticuerpos antigangliósido GQ1b, siendo los siguientes detectados en frecuencia los Ac anti-GM1, GD1a o GalN. Sin embargo, la normalidad de las pruebas de imagen, el LCR, las neurofisiológicas y/o el estudio de autoanticuerpos no descartan la existencia de la EB, por lo que el diagnóstico sigue siendo clínico2,6,8-12.
La definición de la EB implica la existencia de oftalmoplejía y ataxia junto a reflejos exaltados o alteración del sensorio. En nuestros pacientes, ninguno de ellos presentó oftalmoplejía en ningún momento pero sí la alteración del sensorio y la ataxia. En los últimos años han surgido nuevas publicaciones en donde se describen presentaciones atípicas de la EB. En dichos casos, los pacientes presentaba clínicamente la alteración de consciencia junto a ataxia pero la oftalmoplejía no estaba presente. Los autores estudiaron los valores de los anticuerpos anti-GQ1b y encontraron aumento de ellos en todos los casos. Por ello algunos autores propugnan que la presencia de afectación del SNC (alteración de la consciencia o coma) junto a afectación de sistema periférico (oftalmoplejía, neuropatía periférica…), aunque no cumpla todos los criterios clínicos es muy indicativo de un cuadro de EB. En nuestros pacientes se realizó un estudio de autoanticuerpos tanto en sangre periférica como en el LCR. En el caso 2 el resultado pudo negativizarse por la inmunoterapia, ya que la muestra para estudio de autoanticuerpos se extrajo 5 días tras el inicio de esta. En el segundo caso el resultado fue negativo, aunque esto no excluye que pudiese existir algún otro autoanticuerpos no estudiado (nuestro laboratorio no incluye el estudio de GalNAac-GD1a). En relación con el tiempo de evolución de la enfermedad, en los casos pediátricos publicados parece que el desarrollo del cuadro se produce de manera aguda y no en varias semanas, como establecen los criterios que definen la EB. Nuestros pacientes desarrollaron la clínica en menos de una semana y, a pesar de no presentar positividad a anticuerpos anti-GQ1b y no cumplir todos los criterios de EB, se consideró una variante atípica de esta7,8,13-17.
El tratamiento de la EB se basa en la inmunoterapia. Se han utilizado diversas pautas de tratamiento como Ig IV, corticoides a altas dosis, la administración combinada de ambos y/o plasmaféresis. Masaki Odaka et al. proponen el tratamiento conjunto con metilprednisolona a altas dosis e inmunoglobulinas dada la asociación de EB con SGB. En los casos pediátricos descritos en la bibliografía no existe una pauta clara de tratamiento, ya que algunos autores lo inician bien con Ig IV o con corticoides a altas dosis, y se realiza tratamiento combinado si la evolución del paciente es tórpida. En nuestros pacientes, en el caso 1, dado que no se había recibido el resultado del estudio de Herpes viridae, se decidió iniciar el tratamiento con Ig IV por presentar el paciente deterioro clínico progresivo. En el caso 2, el paciente se orientó inicialmente como un posible caso de EMAD. Dado el agravamiento tan severo de la clínica, se decidió iniciar tratamiento con corticoides a altas dosis. Al recibir el resultado de la RM y la evolución tórpida del cuadro, se asoció finalmente tratamiento con Ig IV2,6,8,9,16,18.
El pronóstico de la enfermedad es variable. La mayor parte de los pacientes presentarán una evolución favorable, con recuperación completa en los siguientes 6 meses. Koga et al. revisaron 704 casos en Japón y describieron un grupo de pacientes con presentación que definieron como con clínica atípica, anticuerpos antigangliósido negativos, lesiones en RM y/o marcada pleocitosis en el LCR. Dichos pacientes parece que presentaron los síntomas más allá de los 6 meses y/o no consiguieron recuperación incompleta. Tan et al. definieron en otro grupo de 81 pacientes, encontrando que la presencia de valores de glucosa en sangre y LCR altos se relacionaban también con peor pronóstico. En nuestro caso, ambos pacientes presentaron una recuperación completa, el primero a las 2 semanas y el segundo en aproximadamente 4 meses tras el comienzo de la enfermedad. El caso 1 presentó una pleocitosis leve con valores de glucosa en el LCR y la sangre normales. El caso 2 presentó una pleocitosis moderada en el LCR junto a hiperglucemia en sangre periférica, con valores normales de glucorraquia (tabla 1)2,9,12,16-20.
La EB es una entidad con base autoinmune infrecuente, especialmente en la infancia. La presentación clínica pediátrica parece ser más rápida que la descrita inicialmente por Bickerstaff. Se han descrito casos de EB clásica con autoanticuerpos negativos y de presentación atípica con aumento de anticuerpos anti-GQ1b. Por ello, a pesar de que en nuestros casos no se detectaron dichos auto anticuerpos por lo indicado previamente, la clínica era muy indicativa, por lo que se indicó la inmunoterapia. La rápida identificación de los casos de EB típicos y atípicos y el inicio de inmunoterapia probablemente condicionen la morbilidad de estos cuadros.
Responsabilidades éticasProtección de personas y animalesLos autores declaran que para esta investigación no se han realizado experimentos en seres humanos ni en animales.
Confidencialidad de los datosLos autores declaran que han seguido los protocolos de su centro de trabajo sobre la publicación de datos de pacientes.
Derecho a la privacidad y consentimiento informadoLos autores han obtenido el consentimiento informado de los pacientes y/o sujetos referidos en el artículo. Este documento obra en poder del autor de correspondencia.
FinanciaciónNo se ha recibido ningún tipo de financiación.
Conflicto de interesesNo existe conflicto de intereses en los autores.
