






Estudiar las características clínicas de la esclerosis tuberosa en pacientes de un hospital pediátrico en Ecuador.
Pacientes y métodosSe realizó una búsqueda de historias clínicas de pacientes con esclerosis tuberosa entre los años 2012 y 2016 en un hospital pediátrico de Ecuador; se seleccionó a 31 pacientes que cumplieron los criterios de diagnóstico definitivo. Se analizaron el diagnóstico inicial, el curso clínico y las manifestaciones clínicas y radiológicas de la TSC en los distintos órganos, con particular interés sobre la patología del sistema nervioso central y su relación con los aspectos neurológicos y neuropsiquiátricos.
ResultadosEl 22,5% de los pacientes tuvo historia familiar de TSC. El 61,3% de los pacientes fue diagnosticado antes de los 2 años de edad. En un 74,1% de pacientes las crisis convulsivas fueron las características clínicas más frecuentes al momento del diagnóstico; de este grupo, el 39,1% presentó epilepsia refractaria. En 21 casos (67,74%) se encontró algún tipo de deterioro cognitivo y en la mitad (51,6%) de los pacientes se evidenció trastornos de conducta, los más frecuentes reportados fueron el trastorno del espectro autista y ansiedad. Con respecto a las afectaciones multisistémicas, se encontraron en el 19,35% de los pacientes alteraciones cardiacas, en el 6,45% alteraciones renales, el 6,45% presentó alteraciones oftalmológicas; las alteraciones dermatológicas estuvieron presentes en el 70,96% de los pacientes.
ConclusionesEsta serie de pacientes representa el primer estudio descriptivo ecuatoriano sobre la TSC. Las manifestaciones más frecuentes encontradas son de tipo neurológico: epilepsia, espectro autista y discapacidad intelectual. Consideramos que el diagnóstico inicial debe ser oportuno para prevenir las comorbilidades asociadas a esta patología.
To study the clinical characteristics of tuberous sclerosis (TSC) in patients of a Pediatric Hospital in Ecuador.
Patients and methodsA search of medical records of patients with tuberous sclerosis was carried out between 2012 and 2016 at the Pediatric Hospital of Ecuador. 31 patients with definitive diagnosis criteria were selected. We analyzed the clinical records, initial diagnosis, the clinical course and the clinical and radiological manifestations of TSC at the different organs, with particular interest in central nervous system disorders and their relationship with neurological and neuropsychiatric aspects.
Results22.5% of patients had a family history of TSC. 61.3% of patients were diagnosed before two years of age. In 74.1% of patients, seizures were the most frequent clinical sign at the time of diagnosis. Of this group, 39.1% had refractory epilepsy. In 21 cases (67.74%), some type of cognitive impairment was found, and half (51.6%) of the patients showed behavioral disorders–with the most frequent reports being autism spectrum disorder and anxiety. Multisystem disorders found were heart (19.35%), renal (6.45%), ophthalmological (6.45%), and skin (70.96%), respectively.
ConclusionsThis series of patients represents the first Ecuadorian descriptive study on TSC. The most frequent manifestations were neurological: epilepsy, autistic spectrum and cognitive disability. We believe that initial diagnosis should be timely made, in order to prevent the comorbidities associated with this pathology.
La esclerosis tuberosa o complejo de esclerosis tuberosa (tuberous sclerosis complex, TSC por sus siglas en inglés) es una enfermedad genética poco común que afecta principalmente al sistema nervioso central. La prevalencia estimada a nivel mundial de TSC es de 6,8-12,4 por 100.000 personas. La incidencia al nacimiento es de 1 por cada 5.800 recién nacidos vivos, no encontrándose diferencias entre sexos o etnias1–3. El TSC es una enfermedad de herencia autosómica dominante y expresividad variable4, aunque en 2 tercios de los pacientes se presenta como mutaciones de novo1. El TSC es una patología genética multisistémica que afecta al sistema nervioso central produciendo tumores benignos tipo túberes, que suelen calcificarse o esclerosarse; no obstante, la piel, los riñones, los ojos, el corazón y los pulmones pueden también estar afectados.
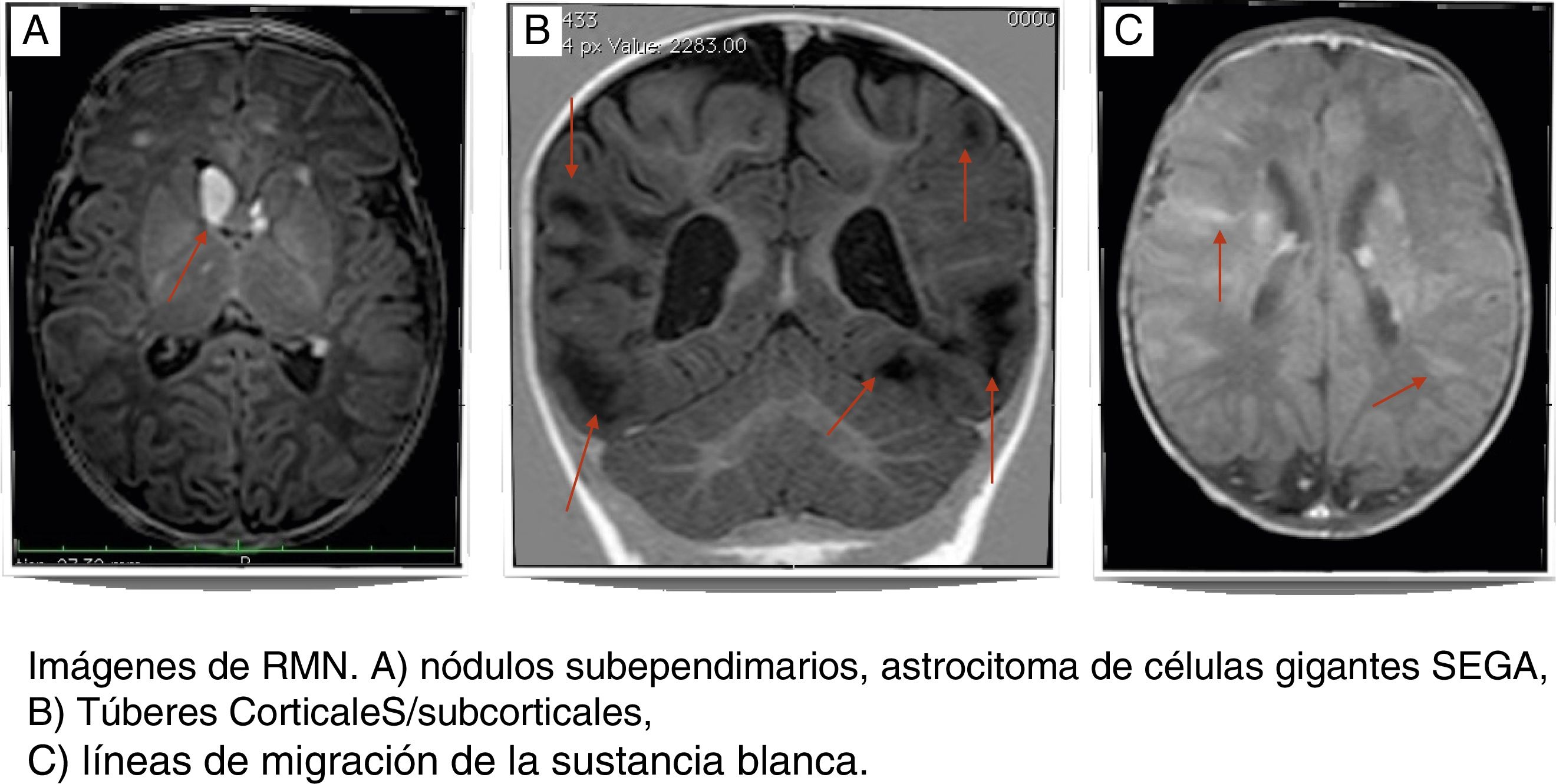
El diagnóstico de TSC suele sospecharse ante la presencia de convulsiones a temprana edad o retraso en el desarrollo. El sistema nervioso central está afectado en el 90% de pacientes con TSC debido a la presencia de lesiones patológicas como túberes corticales o subcorticales, nódulos subependimarios, astrocitomas de células gigantes, líneas de migración de la sustancia blanca (fig. 1). Estas lesiones estructurales suelen estar asociadas a alteraciones neurológicas, como epilepsia y trastornos del espectro autista y el retraso mental2-5. La tomografía computarizada (TC) o una resonancia magnética (RM) permiten la identificación de las lesiones. El ultrasonido del corazón, el hígado y los riñones puede mostrar tumores en esos órganos. Al ser una afectación multisistémica, el TSC se presenta con una variedad de síntomas que dependen del tamaño y la ubicación de los hamartomas, como por ejemplo, convulsiones, autismo, pérdida visual, falla renal y arritmias cardiacas2,4,6. Las 2 lesiones patológicas más frecuentemente encontradas en los niños son los rabdomiomas cardiacos y túberes corticales en el cerebro7,8. Actualmente, el criterio genético es la clave para el diagnóstico de TSC, aunque del 10-25% de los pacientes puede no presentar mutaciones identificables2,6. En países donde el análisis genético aún no es posible, los criterios clínicos siguen siendo la base del diagnóstico.

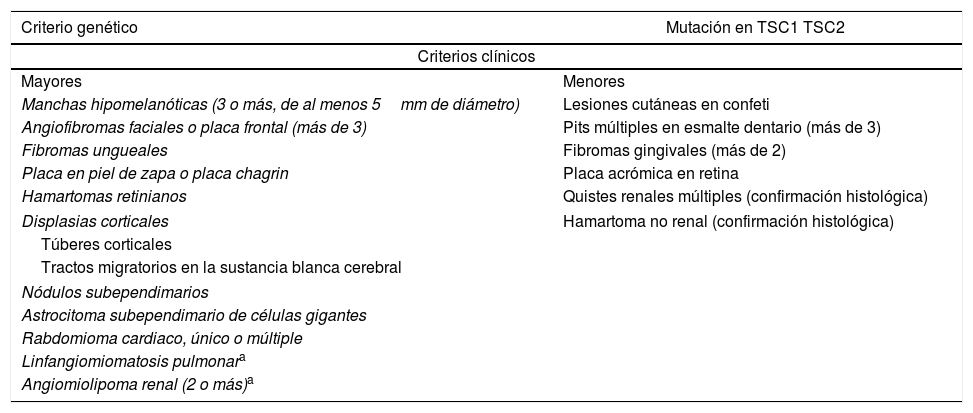
Los criterios diagnósticos fueron revisados por el Consenso Internacional en 2012 (tabla 1)9,10. El diagnóstico definitivo de TSC se establece al detectar 2 criterios mayores, o un criterio mayor con 2 o más criterios menores. Un diagnóstico probable de TSC se realiza ante la presencia de un solo criterio mayor, o en caso de presentarse 2 o más criterios menores2.
Criterios diagnósticos de esclerosis tuberosa
| Criterio genético | Mutación en TSC1 TSC2 |
|---|---|
| Criterios clínicos | |
| Mayores | Menores |
| Manchas hipomelanóticas (3 o más, de al menos 5mm de diámetro) | Lesiones cutáneas en confeti |
| Angiofibromas faciales o placa frontal (más de 3) | Pits múltiples en esmalte dentario (más de 3) |
| Fibromas ungueales | Fibromas gingivales (más de 2) |
| Placa en piel de zapa o placa chagrin | Placa acrómica en retina |
| Hamartomas retinianos | Quistes renales múltiples (confirmación histológica) |
| Displasias corticales | Hamartoma no renal (confirmación histológica) |
| Túberes corticales | |
| Tractos migratorios en la sustancia blanca cerebral | |
| Nódulos subependimarios | |
| Astrocitoma subependimario de células gigantes | |
| Rabdomioma cardiaco, único o múltiple | |
| Linfangiomiomatosis pulmonara | |
| Angiomiolipoma renal (2 o más)a | |
El objetivo del estudio fue describir las características de los pacientes con diagnóstico de TSC en un hospital pediátrico en Ecuador, en un periodo de 5 años (2012-2016). Se incluyó a pacientes con un diagnóstico definitivo de TSC, según criterios del Consenso Internacional del 2012. Los datos demográficos y los registros clínicos de los pacientes se ingresaron en una base de datos Microsoft Excel 2012. Se describen además las alteraciones sistémicas de los participantes, con particular interés sobre el sistema nervioso central en los aspectos neurológicos y neuropsiquiátricos.
ResultadosSe identificó a 31 pacientes con diagnóstico definitivo de TCS, 16 pacientes de sexo femenino y 15 pacientes de sexo masculino. La edad media fue de 5 meses, con un rango entre 0 y 144 meses de edad. El 22,5% de los pacientes tuvieron antecedentes familiares de TSC. En el 61,3% el diagnóstico fue realizado antes de los 2 años de edad y en el 41,9% entre el primer y el sexto mes de vida (media: 16,57 meses). Un caso fue diagnosticado durante la vida intrauterina.
El signo clínico inicial más frecuente de TCS fue la detección de manchas hipopigmentadas (n=8, 25,9%); mientras que el síntoma clínico más frecuente al momento del diagnóstico fue convulsiones (n=23, 74,1%). Del grupo de 23 pacientes con convulsiones, 14 (60,8%) presentaron convulsiones de fácil control farmacológico; sin embargo, 9 (39,1%) presentaron crisis diarias y refractarias al tratamiento. Además, 2 pacientes con criterios de epilepsia refractaria candidatos a resolución quirúrgica fueron intervenidos con callosotomías; uno de los pacientes presentó una disminución significativa de sus crisis convulsivas, mientras que el otro no. El paciente que no respondió al tratamiento quirúrgico presentaba además atrofia córtico-subcortical y múltiples túberes diagnosticados por RM.
El 67,74% de los pacientes tuvieron hallazgos compatibles de TCS en la neuroimagen (TC o RM); entre ellos, los hallazgos más frecuentes fueron: calcificaciones o nódulos subependimarios periventriculares observados en la TC craneal (90%) y túberes corticales o subcorticales en la RM (57,3%); la localización más habitual fue en regiones frontoparietales. Ocho (25,8%) pacientes, a pesar de presentar alteraciones en la neuroimagen, no presentaron crisis convulsivas.
En 21 casos (67,74%) se presentó algún tipo de deterioro cognitivo, 33,33% en forma leve, 38,09% moderado y 29,58% de tipo severo. El 51,6% de los pacientes tuvo un trastorno de conducta, hiperactividad en el 6,25%, desorden de ansiedad en un 62,5% y espectro autista en un 31,25%.
En el grupo de pacientes con convulsiones, el 73,9% presentó algún tipo de deterioro cognitivo y el 56,5% presentó algún tipo de trastorno de conducta (tabla 2). Las alteraciones multisistémicas se resumen en la tabla 3.
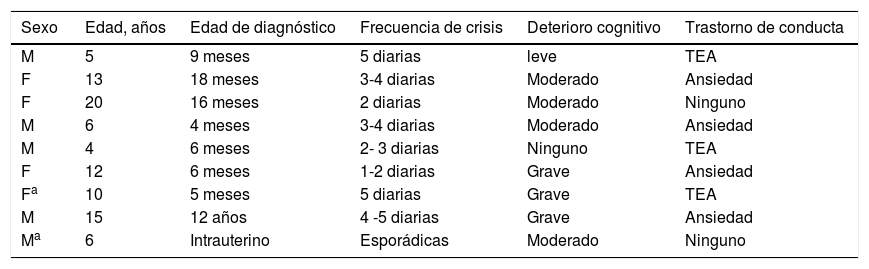
Relación edad de inicio de las crisis, refractariedad, deterioro cognitivo y trastornos de conducta
| Sexo | Edad, años | Edad de diagnóstico | Frecuencia de crisis | Deterioro cognitivo | Trastorno de conducta |
|---|---|---|---|---|---|
| M | 5 | 9 meses | 5 diarias | leve | TEA |
| F | 13 | 18 meses | 3-4 diarias | Moderado | Ansiedad |
| F | 20 | 16 meses | 2 diarias | Moderado | Ninguno |
| M | 6 | 4 meses | 3-4 diarias | Moderado | Ansiedad |
| M | 4 | 6 meses | 2- 3 diarias | Ninguno | TEA |
| F | 12 | 6 meses | 1-2 diarias | Grave | Ansiedad |
| Fa | 10 | 5 meses | 5 diarias | Grave | TEA |
| M | 15 | 12 años | 4 -5 diarias | Grave | Ansiedad |
| Ma | 6 | Intrauterino | Esporádicas | Moderado | Ninguno |
F: femenino; M: Masculino; TEA: trastorno de espectro autista.
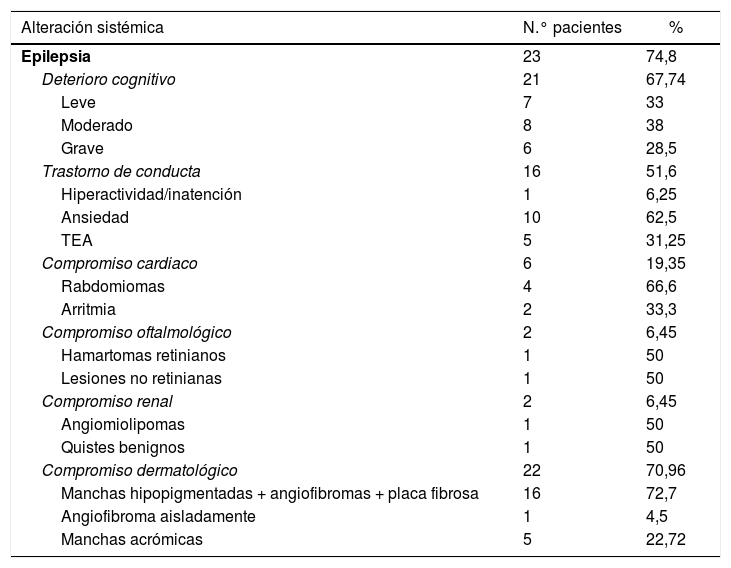
Alteraciones multisistémicas en el grupo de estudio
| Alteración sistémica | N.° pacientes | % |
|---|---|---|
| Epilepsia | 23 | 74,8 |
| Deterioro cognitivo | 21 | 67,74 |
| Leve | 7 | 33 |
| Moderado | 8 | 38 |
| Grave | 6 | 28,5 |
| Trastorno de conducta | 16 | 51,6 |
| Hiperactividad/inatención | 1 | 6,25 |
| Ansiedad | 10 | 62,5 |
| TEA | 5 | 31,25 |
| Compromiso cardiaco | 6 | 19,35 |
| Rabdomiomas | 4 | 66,6 |
| Arritmia | 2 | 33,3 |
| Compromiso oftalmológico | 2 | 6,45 |
| Hamartomas retinianos | 1 | 50 |
| Lesiones no retinianas | 1 | 50 |
| Compromiso renal | 2 | 6,45 |
| Angiomiolipomas | 1 | 50 |
| Quistes benignos | 1 | 50 |
| Compromiso dermatológico | 22 | 70,96 |
| Manchas hipopigmentadas + angiofibromas + placa fibrosa | 16 | 72,7 |
| Angiofibroma aisladamente | 1 | 4,5 |
| Manchas acrómicas | 5 | 22,72 |
El TSC se produce debido a mutaciones en 2 genes diferentes. Los genes implicados son el TSC1 (gen de la hamartina, cromosoma 9q34) y el TSC2 (gen de la tuberina, cromosoma 16p13.3); ambas proteínas forman un complejo tuberina-hamartina que actúa inhibiendo la actividad de la vía mTOR (diana de rapamicina en células de mamífero), la cual regula la síntesis proteica, la proliferación, el crecimiento, el metabolismo, la supervivencia de las células y la plasticidad sináptica2,4,11. Los datos encontrados en este estudio confirman que entre el 65 y 75% de los casos no suelen tener historia familiar de TCS, y que la enfermedad se produce como consecuencia de mutaciones de novo3,4; en nuestra muestra, solo el 22,78% tuvo antecedentes familiares de TSC.
Las mutaciones en cada uno de los genes se asocian a diferentes características de la patología. La mutación del gen TSC1 está asociada a una expresión menos grave de la enfermedad pero a una mayor incidencia familiar; mientras que la mutación del gen TSC2 se asocia a formas espontáneas pero más graves de la enfermedad1,12. La mutación TSC2 además se relaciona con una presentación temprana epilepsia, una mayor incidencia de refractariedad, un menor cociente intelectual y un mayor número de túberes1,12,13.
Los túberes son las malformaciones focales más frecuentes del desarrollo cortical (fig. 1). Los túberes se encuentran en el 80% de los pacientes con TSC, se desarrollan debido a la proliferación descontrolada de células gliales y neuronas, y se ubican generalmente en la unión de la sustancia gris y blanca, especialmente en la región frontal5,8,12. Los túberes proliferan desde la séptima a la duodécima semana de edad gestacional y pueden ser diagnosticados intraútero mediante RM fetal4,14. En nuestra muestra, se identificaron túberes en el 57,34% de los casos, frecuentemente en regiones frontoparietales; un solo un caso fue diagnosticado durante la vida intrauterina.
Los nódulos subependimarios son lesiones que se presentan hasta en el 80% de los individuos con TSC y se localizan en las paredes de los ventrículos laterales y del tercer ventrículo, generalmente son asintomáticos y se calcifican a lo largo de la vida5,8. Los nódulos ependimarios pueden evolucionar a un astrocitoma de células gigantes (SEGA), en un 6 al 18% de los casos (fig. 1); los SEGA generalmente son unilaterales y se sitúan en las inmediaciones de los agujeros de Monro5,15. La transformación a SEGA ocurre en nódulos mayores de 5mm no calcificados y que se realzan con gadolinio en la RM craneal. La conversión a un SEGA suele ser un proceso gradual con mayor probabilidad de que ocurra en las 2 primeras décadas de la vida2,4,8. No se encontraron pacientes con SEGA; sin embargo, la presencia de nódulos subependimarios se presentó en el 90% de los pacientes de este estudio.
Las manifestaciones más frecuentes en niños con TSC son de tipo neurológico, hasta el 85% de los casos se presentan como epilepsia, espectro autista, discapacidad intelectual y, por su gravedad, constituyen la principal causa de morbimortalidad6,16. Entre el 72 y 85% de los individuos con TSC tienen historia de crisis convulsivas y en el 80% de los casos empiezan antes de los 3 años de vida. Estos datos se asemejan a los encontrados en nuestra muestra; el 74,8% presentó crisis convulsivas como síntoma inicial y en el 61,25% las crisis empezaron antes de los 2 años de edad. El tipo de crisis convulsiva más frecuentemente reportado son de espasmos infantiles o crisis parciales motoras, o un conjunto de las 2, aunque otro tipo de crisis convulsivas también pueden presentarse6,13,17. Las convulsiones pueden tener un origen focal o multifocal, que suele tener correspondencia eléctrica y topográfica en el electroencefalograma (EEG) y con los túberes corticales en la RM, demostrando su potencial papel como zona epileptógena12,18. El proceso de epileptogenésis que ocurre en el TSC se produce debido cambios en la expresión de receptores GABA y glutamato en las células gigantes y neuronas displásicas; el déficit de interneuronas gabaérgicas explicaría el inicio precoz de las crisis y a la gran refractariedad al tratamiento farmacológico18,19, lo cual repercute negativamente en el desarrollo cognitivo del niño12,13,20. En nuestra serie, el 34,7% de los pacientes presentaron epilepsias refractarias con una mayor frecuencia de deterioro cognitivo moderado a severo, además de varios trastornos de conducta (tabla 4).
Características de los pacientes
| Característica | N (%) |
|---|---|
| Sexo | |
| Femenino | 16 (51,6) |
| Masculino | 15 (48,3) |
| Edad media | 5 meses (0-144 meses) |
| Antecedentes | |
| Con antecedentes familiares | 7 (22,5) |
| Sin antecedentes familiares | 24 (77,4) |
| Edad de diagnóstico | |
| Antes 2 años | 19 (61,3) |
| Después 2 años | 12 (38,7) |
| Signo clínico inicial | |
| Crisis convulsivas | 23 (74,1) |
| Manchas hipopigmentadas | 8 (25,9) |
| Crisis convulsivas | |
| Crisis refractarias | 9 (39,1) |
| Crisis controladas | 14 (60,8) |
| Hallazgos de neuroimagen | |
| Positivo | 21 (67,7) |
| Negativo | 10 (32,2) |
| Deterioro cognitivo | |
| Positivo | 21 (67,7) |
| Leve | 7 (33) |
| Moderado | 8 (38) |
| Severo | 6 (29) |
| Negativo | 10 (32,2) |
| Trastorno de conducta | |
| Presente | 16 (51,6) |
| Ausente | 15 (48,3) |
Se han definido los principales factores de riesgo para un peor pronóstico en el desarrollo cognitivo, como el inicio de crisis convulsivas en el primer año de edad, la presencia de espasmos infantiles, la epilepsia refractaria, la mutación en el gen TSC2 y la presencia de más de 7 túberes8,13. Algunos autores recomiendan el inicio precoz del tratamiento antiepiléptico en pacientes en los que en el EEG se evidencie descargas paroxísticas, incluso antes de la aparición de crisis clínicas, ya que de esta manera se podría prevenir los efectos de la epileptogénesis, con la aparición de crisis refractarias que provocan un efecto deletéreo en la cognición y conducta del individuo16,21. En nuestros datos se encontró un 67,74% de deterioro cognitivo. La literatura reporta que entre el 60 y 70% de los pacientes tiene una inteligencia normal, con déficits cognitivos leves, pero entre el 30 y el 40% puede presentar un retraso mental profundo20,22.
El trastorno del espectro autista se identificó en el 31,25% y el desorden de ansiedad en el 62,5% de nuestra muestra; en la literatura, estas alteraciones se han relacionado con mayor frecuencia con pacientes con mutaciones en TSC2 y con presencia de túberes corticales frontales y temporales, lo que provocaría una temprana disfunción de áreas asociativas12,22,23.
Las afectaciones multisistémicas reportadas con mayor frecuencia en nuestra muestra fueron alteraciones cardiacas en el 19,35% y alteraciones renales y oftalmológicas en el 6,45%, mientras que las alteraciones dermatológicas se presentaron en el 70,96% de los pacientes.
Dado que en algunos pacientes existió un seguimiento del cuadro clínico, enfocado únicamente a los síntomas o signos iniciales, presentamos un posible esquema de diagnóstico y seguimiento que facilite la práctica clínica y el manejo a largo plazo de los pacientes (tabla 5)1,6,8,24. Finalmente, consideramos que la valoración de estos pacientes debe ser integral, multidisciplinar, temprana y efectiva, ya que, podría prevenir comorbilidades asociadas y mejorar el pronóstico.
Diagnóstico y seguimiento de pacientes con TSC
| Diagnóstico inicial | Seguimiento | |
|---|---|---|
| Sintomático | Asintomático | |
| Fondo de ojo | + | – |
| RM cerebral o TC cerebral | Según clínica | Cada 1 a 3 años |
| EEG, si sospecha de convulsiones | Según clínica | – |
| ECO y ECG cardiaco | Cada 6 meses a 1 año hasta ver involución o estabilización | Cada 3 años Hasta adolescencia |
| ECO renal | Cada 6 meses a 1 año hasta ver involución o estabilización | Cada 3 años Hasta adolescencia |
| Evaluación dermatológica | + | – |
| Valoración desarrollo neurológico | + | Inicio de escolarización |
Adaptado de Sanz24.
Esta serie de pacientes con TSC representa el primer estudio descriptivo en Ecuador; no obstante, a pesar de pequeñas diferencias identificadas, nuestro trabajo apoya lo publicado y fomenta la realización de nuevos estudios en esta línea de investigación.
FinanciaciónFinanciado por autores.
Conflicto de interesesLos autores del presente manuscrito declaran no tener conflicto de interés con el tema en referencia.
